
INTRODUCTION
Splicing, which involves excising introns from pre-mRNA and then ligating the exons (De Conti et al., 2013), occurs in a wide range of eukaryotes, including yeast, and is mediated by the spliceosome, a complex assembly of multiple snRNPs and proteins (Karijolich & Yu, 2010). Splicing is crucial for the production of mature mRNA, which is translated into proteins, and for the maintenance of protein functional diversity (Singh, 2002). Consequently, splicing aberrations can be significantly detrimental to cellular function (Choi et al., 2023; Lee et al., 2023).
The distinct sequence motifs, 5’ splice site, branch site, and 3’ splice site, mark intron–exon junctions. The branch site, which is positioned slightly closer to the intron’s 3’ end, is typically followed by a polypyrimidine tract, with the branch being centered around an adenine nucleotide (Xie et al., 2023). A conserved GU sequence characterizes the 5’ splice site, and an AG sequence marks the 3’ splice site (Kitamura-Abe et al., 2004).
Compared with other eukaryotes, yeast has fewer intron-containing genes (Stajich et al., 2007), and most, such as ribosomal protein (RP) genes, contain a single intron and are often highly expressed. However, intron numbers generally increase with increasing organism complexity. For instance, approximately 10% of all human genes contain exons (Sakharkar et al., 2004). Therefore, splicing must be tightly regulated.
In the budding yeast, Saccharomyces cerevisiae, Small Ubiquitin-like Modifier (SUMO) proteins are conjugated to target proteins through the sequential action of E1 activating enzymes, E2 conjugating enzymes, and E3 ligases (Melchior, 2000). This post-translational modification plays pivotal roles in various cellular processes, including transcriptional regulation, DNA repair, translation, and cell cycle progression (Choi et al., 2021; Ryu and Hochstrasser, 2021; Ryu, 2022). A subset of SUMO conjugates undergoes deSUMOylation catalyzed by the proteases, Ulp1 and Ulp2 (Ryu et al., 2019).
Recent studies indicate that for efficient pre-mRNA splicing, SUMOylation is significantly involved in spliceosomal protein regulation (Pozzi et al., 2017). For example, SUMOylation at specific sites, such as Lys-289 and Lys-559 on PRP3, a component of the U4/U6-U5 snRNP complex, is critical. Mutations that disrupt these SUMOylation sites are reported to impede recruitment to the active spliceosome, highlighting the importance of precise SUMOylation for RNA processing dynamics. In vitro, recombinant SENP1, a SUMO protease, is reported to diminish pre-mRNA splicing efficiency (Pozzi et al., 2017). Moreover, in the human cell line, HeLa, mRNA splicing-related proteins are known to undergo endogenous polySUMOylation (Bruderer et al., 2011). Here, we investigated the role of Ulp2 protein, a protease involved in SUMOylation regulation, in RNA splicing.
MATERIALS AND METHODS
Table 1 shows the yeast strains used in this study. To generate HYS536, the snt309Δ::KanMX4 cassette was PCR-amplified using SNT309 KO primers and HYS418. The amplified products were used to transform MHY500, followed by transformant selection on YPD+G418. The cells were grown at 30°C in a YPD medium with appropriate supplements. RNA was isolated from cells (one OD 600 equivalent) grown to the mid-exponential phase.
| Strain | Genotype | Source |
|---|---|---|
| HYS114 (WT) | MATa his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 gal2 | This study |
| MHY1379 | MATa his3-Δ200 leu2-3,112 lys2-801 trp1-1 ura3-52 ulp2Δ::HIS3 YCplac33-ULP2 | (Li & Hochstrasser, 2000) |
| HYS418 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 snt309Δ::KanMX4 | TAP tag library |
| HYS536 (snt309Δ) | MATa his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 gal2 snt309Δ::KanMX4 | This study |
| HYS93 (ubc9-1) | MATa his3-Δ200 leu2-3,112::LEU2::ubc9-1 ura3-52 lys2-801 trp1-1 gal2 ubc9Δ::TRP1 | This study |
| HYS90 (ulp1ts) | MATa his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 ulp1Δ::HIS3 yCplac22-ulp1ts(3-33) | This study |
| HYS183 (ulp2Δ) | MATa his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 ulp2Δ::HIS3 | This study |
| MHY7863 | MATa his3-Δ200 leu2-3,112 lys2-801 trp1-1 ura3-52 ULP2-6xGly-3xFlag::HIS3MX6 | (Ryu et al., 2016) |
WT, wild-type.
The RNA-seq dataset, GSE121898, from Gene Expression Omnibus, was used to re-analyze ulp2Δ’s differentially expressed introns compared with the wild-type (WT) (Ryu et al., 2018). Volcano plots were drawn using R studio’s ggplot2 package.
The ChIP-seq dataset, GSE130623 (Ryu et al., 2019), from Gene Expression Omnibus, was used for the re-analysis of Ulp2-Flag enrichment at RP introns. ChIP-seq tracks were viewed using the Integrative Genomics Viewer (https://igv.org/) (Liu et al., 2024).
Total RNA was extracted from samples using an APure™ total RNA kit (AP Bio, Brooklyn, NY, USA). Next, 1 mg of RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Table 2 shows the sequences of the oligonucleotides used for qPCR. For qPCR, to determine the expression of the RP gene, RPL31B, cDNA was diluted at 1:100. All qPCR reactions were done in technical triplicate, and relative RNA levels were determined using the comparative Ct (ΔΔCt) method (Schmittgen & Livak, 2008; Ryu et al., 2020b).
RT-PCR analyses were done four times. Splicing efficiency was compared using a Student two-tailed t-test. Data are presented as mean±SEM. p<0.05 indicates statistically significant differences.
Gene ontology (GO) analysis was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID). GO data were filtered by EASE score, a modified Fisher’s exact p-value used on the DAVID database, with an EASE score of <0.1. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was conducted using the YeastErichr database, and the data were filtered based on a Fisher’s exact test with an adjusted p-value of <0.05 and a combined score.
RESULTS
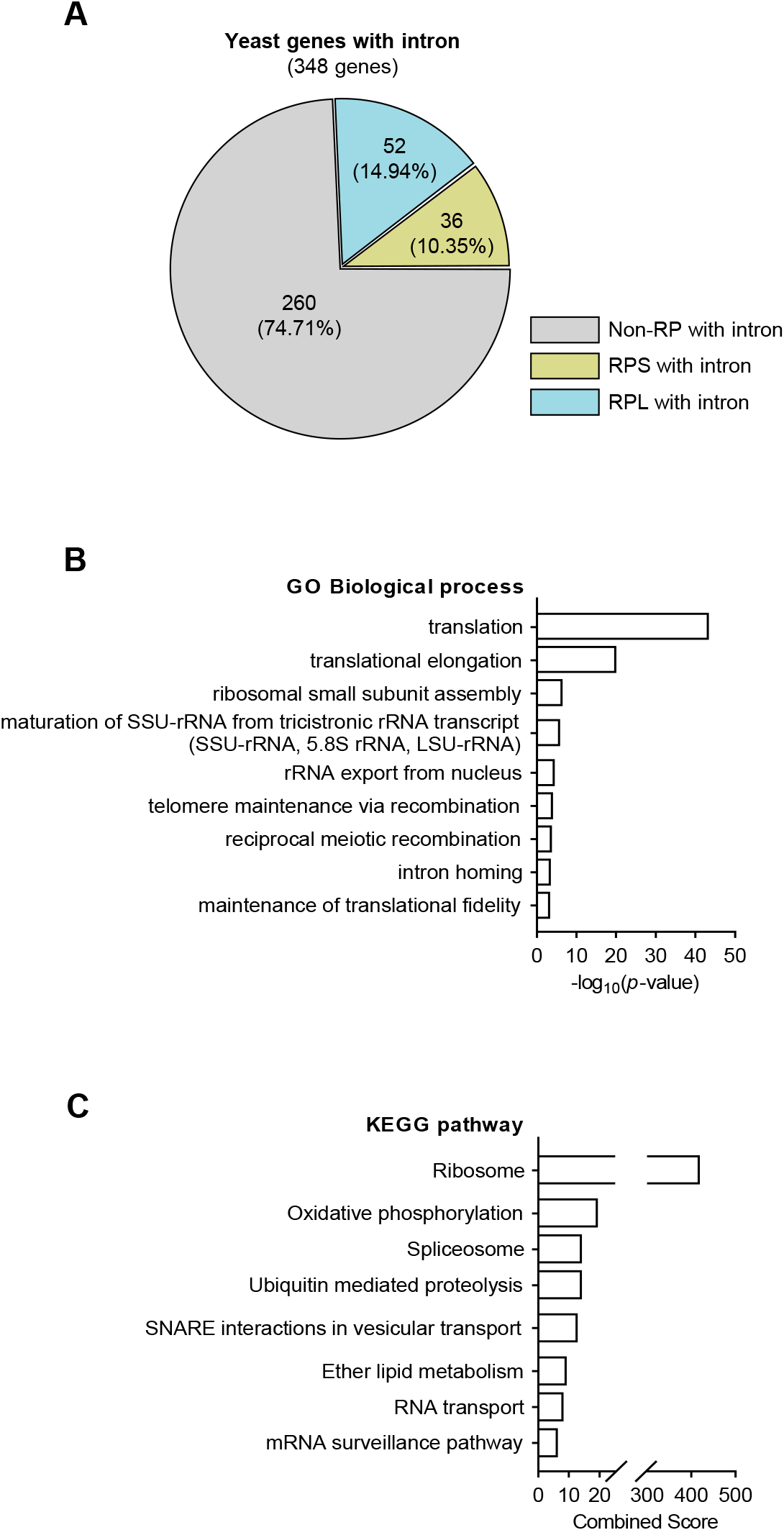
Unlike other eukaryotes, yeast exhibits a relatively modest number of intron-containing genes. Here, using the Saccharomyces Genome Database to determine the number of yeast genes with introns, we identified 348 budding yeast intron-bearing genes (Fig. 1A). The genes were categorized into the ribosomal and non-ribosomal groups, with further classification into the large and small ribosomal subunit genes, and 25% of the intron-containing genes were confirmed to be RP genes. Next, we used GO and KEGG pathway analyses to determine the functional categories of intron-containing genes (Fig. 1B–C). These analyses revealed that RP genes essential for translation were significantly abundant, suggesting that during post-transcriptional modification, many RP gene intronic regions are accurately regulated through splicing.
Because recent studies indicate that spliceosomal protein SUMOylation regulates splicing (Pozzi et al., 2017), we investigated how SUMOylation deregulation in a ULP2-deficient strain impacts splicing. Next, we used a previously reported RNA-seq WT and ulp2Δ mutant dataset (GSE121898) to re-identify differentially expressed introns in the intron-containing genes as depicted in Fig. 1 (Fig. 2A). When compared with the WT, this analysis identified significant changes in RNA transcript levels in the ulp2Δ mutants’ intronic regions (Fig. 2B). Notably, some RP transcripts had more than a twofold expression increase in ulp2Δ mutants’ intronic regions (Table 3). Among some RP genes, we observed an increase in the levels of RNA transcripts that retained introns within the entire set of intron-containing RPs (Fig. 2C), suggesting that ULP2 deletion can dysregulate RNA splicing.
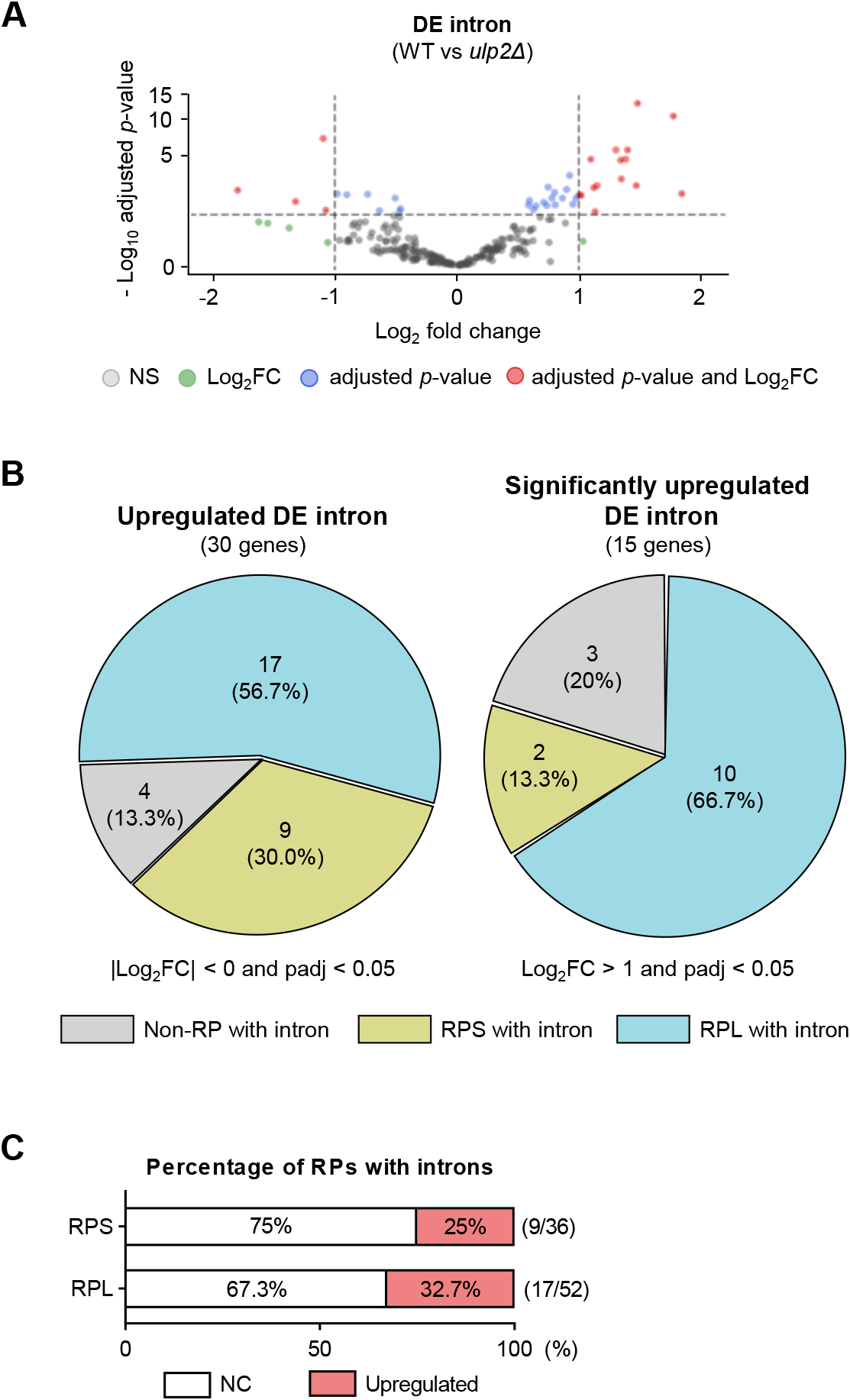
RP, ribosomal protein; WT, wild-type.
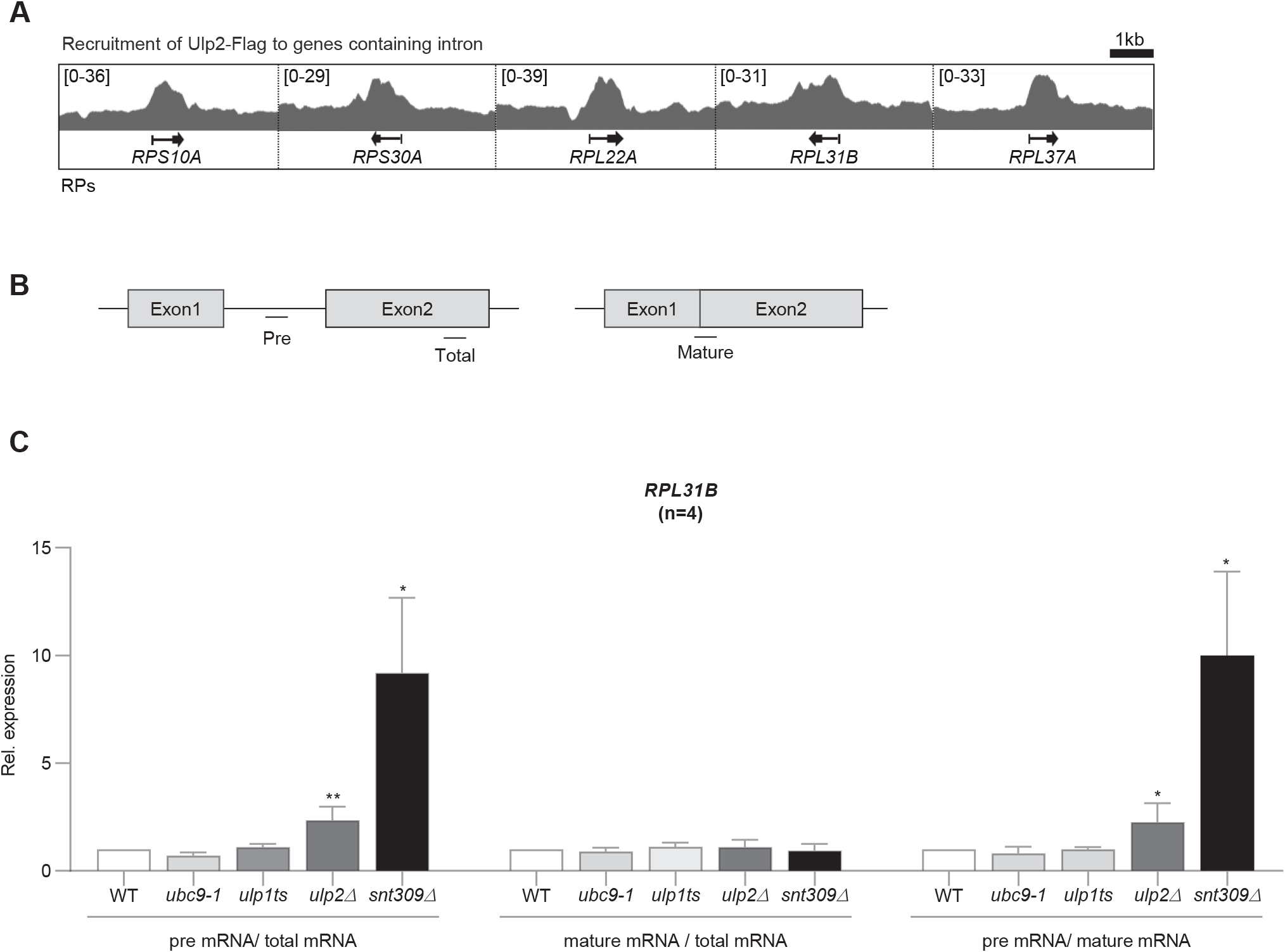
To confirm if the differences observed from the RNA-seq results are actually because of Ulp2 binding, we investigated Ulp2 protein binding to each RP intron using a previously reported ChIP-seq dataset (GSE130623) based on Ulp2-Flag. This analysis confirmed that Ulp2 is highly bound to RP intronic regions (Fig. 3A). Based on these results, to examine RPL31B’s splicing efficiency using SUMO pathway mutants, we designed RT-PCR primers divided into three parts (Fig. 3B). By designing primers within the RPL31B’s intron, we could measure pre-mRNA amounts, and primers targeting the two exons that are joined through splicing allowed the assessment of mature mRNA levels. As a control, SNT309, an mRNA-splicing factor was knocked out (Albulescu et al., 2012). For SUMO pathway mutants, we impaired SUMO-E2 conjugating enzyme function with ubc9-1, and functionally impaired or deleted SUMO proteases with ulp1ts and ulp2Δ. RNA was then extracted from each strain, followed by RT-PCR analysis of the splicing efficiency of RPL31B transcripts (Fig. 3C). This analysis revealed that although pre-mRNA levels were not as high as in the snt309Δ mutant, they were more than twice higher than in the WT, indicating that Ulp2 is required for splicing of some RP transcript.
DISCUSSION
Although recent studies indicate that SUMOylation regulates various RNA metabolism stages that are associated with tumorigenesis and cancer progression (Ryu et al., 2020a; Cao et al., 2023), it is unclear if the SUMO proteases that regulate SUMOylation directly impact RNA splicing. Here, we show that the Ulp2 protein affects the splicing process. These study’s results suggest that Ulp2 protein can regulate several SUMOylated proteins involved in splicing, which may affect splicing efficiency. Additionally, we show that during post-transcriptional modification, ribosomes, which play a crucial role in protein synthesis, can be partially regulated by the Ulp2 protein. In future studies, we will aim to provide mechanistic insights into Ulp2 function in post-transcriptional modification.