





INTRODUCTION
The luteinizing hormone receptor (LHR) is a member of the family of the 7 transmembrane G protein-coupled receptors (GPCRs), one of the largest gene families (Kudo et al., 1996). In particular, the LHR and follicle-stimulating hormone receptors (FSHRs) form a subgroup of the glycoprotein hormone receptors (Segaloff & Ascoli, 1993; Ascoli et al., 2002). The LH receptor (LHR) gene has been associated with an abundance of naturally occurring mutations related to reproductive failures in mammals (Meehan & Narayan, 2007; McGee & Narayan, 2013). Many naturally occurring constitutively activating/or inactivating mutations of both human LHR (hLHR) and hFSHR have been described (Kraaij et al., 1995; Tao et al., 2000; Wu et al., 2000; Tao, 2006; Althumairy et al., 2020).
The first mutation reported is hLHR-D578Y (equivalent to D590Y in eel LHR) in transmembrane VI (Shenker et al., 1993). Since the original report of D578Y, additional mutations have been reported, including M398T (equivalent to M410T in eel LHR) in located in the second transmembrane helix (Ignacak et al., 2000), L457R (equivalent to L469R in eel LHR) in the transmembrane III (Latronico et al., 1998). The hLHR-L457R mutant was first identified on the basis that cells expressing this mutant displayed remarkably basal cyclic adenosine monophosphate (cAMP) levels. However, cells expressing the L457R mutant were unresponsive to further hormonal stimulation (Zhang et al., 2005 , 2007; Latronico & Segaloff, 2007). The rat LHR (rLHR)-D556H mutation (rLHR-D556H; equivalent to the hLHR-D578H mutation) has also been shown to result in a marked increase in the basal cAMP response (Meehan & Narayan, 2007). Although the inactivating rLHR-D383N (equivalent to D417N in eel LHR) and -R442H mutations have been reported to not affect the binding of the human chorionic gonadotropin (hCG), they do impair signal transduction (Dhanwada et al., 1996). We have also previously reported that the rLHR-D383N and Y524F (equivalent to Y558F in eel LHR) mutations were found to be signal impairing mutations (Min et al., 1998).
Recent research studies from our laboratory have elucidated the signal transduction of the eel LHR (Byambaragchaa et al., 2018) and eel FSHR (Kim et al., 2016, 2019) on deglycosylated ligands. In recently, we also first reported in fish species that eel FSHR-D540G mutant exhibited a highly increase in the basal cAMP response (Byambaragchaa et al., 2020). Although the activation effects of these mutants have been relatively well demonstrated in hLHR, very less is known about the signal transduction leading to the activation and inactivation in fish LHR.
Thus, this study aimed to determine these mechanisms by analyzing three constitutively activating (M410T, L469R, and D590Y in eel LHR), and two inactivating (D417N and Y558F in eel LHR) mutations known as highly conserved regions of transmembrane domains among the mammalian LHRs. Here, we report that the basal cAMP response was constitutively involved in activating eel LHRs, but the inactive mutants impaired the signal transduction.
MATERIALS AND METHODS
Polymerase chain reaction (PCR) reagents and endonucleases were purchased from Takara (Osaka, Japan). Oligonucleotides were synthesized by Genotech (Dajeon, Korea). The pcDNA3 mammalian expression vector, Chinese hamster ovary (CHO)-suspension (CHO-S) cells, MAX transfection reagent, Lipofectamine™-3000, Freestyle CHO medium, and antibiotics (penicillin and streptomycin) were obtained from Invitrogen (Carlsbad, CA, USA). The pGEM-T easy cloning vector was purchased from Promega (Madison, WI, USA). CHO-K1 cells were obtained from the Japanese Cancer Research Resources Bank ( JCRB, Tokyo, Japan). A homogeneous time-resolved fluorescence (HTRF) cAMP assay kit was purchased from Cisbio (Codolet, France). Monoclonal antibodies (5A11, 11A8, and 14F5) and rec-eel LH from CHO-K1 cells were produced in our lab, as previously reported (Kim et al., 2016). The horseradish peroxidase (HRP) labeling of the 8A11 monoclonal antibody was generously performed by Medexx (Seongnam, Korea). Eel LHR cDNA was cloned from eel ovaries and testes, as previously reported (Byambaragchaa et al., 2020). QIAprep-Spin plasmid kits were purchased from Qiagen (Hilden, Germany). Disposable spinner flasks were purchased from Corning (Corning, NY, USA). Centrifugal Filter Devices were purchased from Amicon Bio (Billerica, MA, USA). All other reagents used in this experiment were obtained from Sigma-Aldrich (St. Louis, MO, USA) or Wako Pure Chemicals (Osaka, Japan). The procedures and protocols used in this study were ethically reviewed and approved in accordance with the guidelines of the Hankyong National University committee (Number: 2018-03-01).
An overlap extension PCR strategy was used to create the activating and inactivating mutants in eel LHR cDNA as described previously (Byambaragchaa et al., 2020). Two different sets of PCR primers were used to amplify each mutant fragment. In step 1, the first set of fragments were amplified using forward and reverse primers (mutation primer). The second set of fragments were then amplified using forward (mutation primer) and reverse primers. In step 2, the amplified fragments (first and second set of fragments) from step 1 were used as templates to amplify the completely mutated fragments. The primer sequences used in these experiments are shown (Table 1). The full-length PCR products synthesized in step 2 were cloned into a pGEM-T easy vector. Plasmids were extracted and sequenced to confirm the presence of the mutations. A schematic representation of the naturally occurring mutation sites for the 3 activating (M410T, L469R, and D590Y) and 2 inactivating (D417N and Y558F) mutants in eel LHR is shown in Fig. 1.
Underlined nucleotides are sites of mutagenesis, while bold nucleotides shown the EcoRI and XhoI restriction sites for cloning into the expression vector.
LHR, luteinizing hormone receptor.
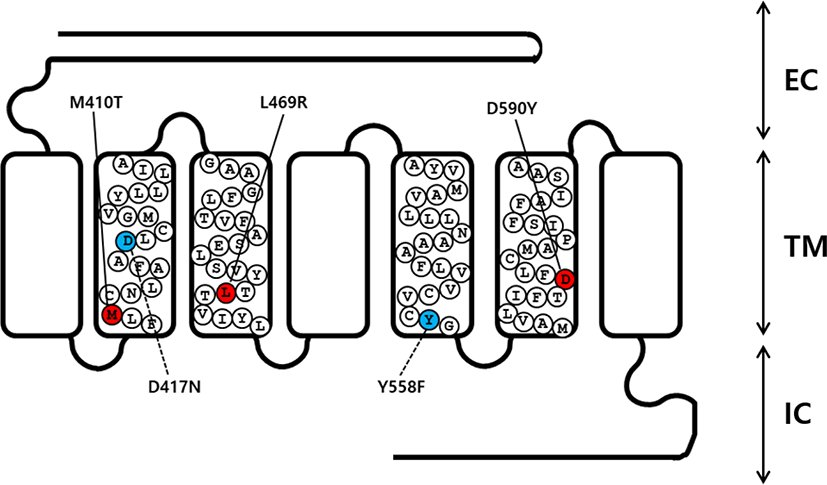
cDNAs encoding wild type (WT) and mutant eel LHR were digested with the Eco RI and Xho I restriction enzymes. The resulting fragments were then ligated into the pcDNA3 and pCORON1000 SP VSV-G expression vector, as previously described (Byambaragchaa et al., 2018). Plasmids were then purified, and the presence of the correct insert was confirmed through analysis with restriction enzymes. Finally, we constructed a total of 6 receptor genes, including WT eel LHR, M410T, L469R, D590Y, D417N, and Y558F.
CHO-K1 cells were cultured in growth medium (Ham’s F-12 medium containing 2 mM glutamine, 50 U/mL penicillin, 50 µg/mL streptomycin, and 10% fetal bovine serum [FBS]). Cells were grown to 80%–90% confluence in 6-well plates followed by transfection with mutant plasmids. CHO growth medium containing 20% FBS was added to each well 5 h after transfection. Cells were used for cAMP analysis at 48 h after transfection.
For ligand production, the rec-eel LH expression vector was transfected into CHO-S cells using the FreeStyle™ MAX reagent transfection method according to the manufacturer’s instructions, and as was previously reported in our lab (Byambaragchaa et al., 2018). On the day of transfection, cell density was approximately 1.2–1.5×106 cells/mL. The FreeStyle™ MAX Reagent and eel LH plasmid were diluted, and gently mixed by inverting the tube. The DNA-FreeStyle™ MAX reagent was incubated for 10 min at 25°C to allow the formation of complexes. The complexes were added to 200 mL of cell-containing medium. Culture media were collected on day 7 after transfection; supernatants were collected and frozen at –80°C.
Rec-eel LH was quantified using a double-sandwich enzyme-linked immunosorbent assay (ELISA) performed in plates coated with the 5A11 monoclonal antibody, as previously described (Kim et al., 2016). A volume of 100 µL of the rec-eel LH sample was added to the wells and then incubated for 1 h at 25°C. After washing 3 times with PBS-T, HRP-conjugated anti-eel11A8 antibody in PBS was added to the plates and they were incubated for 1 h at 25°C. After washing, wells were incubated with 100 µL of substrate solution for 20 min at 25°C. The reaction was stopped by adding stop solution (50 µL of 1 M H2SO4). Absorbance at 450 nm was measured in each well using a microplate reader Cytation 3 (BioTek, Winooski, VT, USA).
The accumulation of cAMP in CHO-K1 cells expressing WT and mutant eel LHRs was measured using cAMP Dynamic 2 assay kits (Cisbio Bioassays, Codolet, France), as previously described (Byambargchaa et al., 2018). Briefly, cells transfected with WT and mutant eel LHRs were added at 10,000 cells per well into a 384-well plate 48 h after transfection. Cells were stimulated by incubation with the agonist for 30 min at 25°C. cAMP level was detected by measuring the decrease in HTRF energy transfer (665 nm/620 nm) using an Artemis K-101 HTRF microplate reader (Kyoritsu Radio, Tokyo, Japan). The specific signal-Delta F (energy transfer) is inversely proportional to the concentration of cAMP in the standard or sample. Results were calculated based on the 665 nm/620 nm ratio and expressed as Delta F% (cAMP inhibition), according to the following equation: [Delta F% = (standard or sample ratio – sample negative) × 100 / ratio negative]. The cAMP concentrations were calculated from the Delta F% values using the Prism software (GraphPad, La Jolla, CA, USA).
The MultAlin interface-multiple sequence alignment software was used for sequencing results. The GraphPad Prism 6.0 was used for the analysis of the production of cAMP and GraFit 5.0 (Erithacus Software Limited, Surrey, UK) was used for the cAMP EC50 values and analyses of stimulation curves. Curves fitted in a triplicate experiment were normalized to the background signal measured for mock-transfected cells. One-way ANOVA and Turkey’s Multiple Comparison tests were used to compare the results between samples, using GraphPad Prism 6.0. Differences were indicated as significant between the groups (p<0.05).
RESULTS
As previously reported, the eel LHR is known to consist of 2115 nucleotides encoding 705 amino acids (Byambaragchaa et al., 2018). In order to generate substitute mutations at target amino acids, we used an overlap extension PCR strategy with primers designed to change target nucleotides. To investigate the effects on the interaction of hormones to the receptor in the eel LHR activation system, we generated 3 constitutively activating mutations in the II, III, and VI transmembrane helices of LHR. These mutant receptors were designated M410T, L469R, and D590Y. We also constructed 2 inactivating mutations of eel LHR, designated as D417N and Y546F in the II and V transmembrane helices, respectively (Fig. 1).
The effects of activating mutations on the basal and eel LH-stimulated cAMP responsiveness are summarized in Fig. 2A. The basal and Rmax cAMP responses in WT receptor were demonstrated to be 1.2 and 87.5 nM/ 104 cells, respectively. The production of cAMP was exhibited an increased in a dose-dependent manner. The EC50 value of the eel LH-stimulated cAMP response was shown to be approximately 18.9 ng/mL. In cells expressing the constitutively activating mutants (M410T, L469R, and D590Y) the induced basal cAMP responsiveness was increased by 4.8±0.3, 22.9±1.5, and 9.3±0.8 ng/104 cells, respectively (Fig. 2B and Table 2). In contrast to CHO-K1 cells harboring the WT receptor, cells expressing the M410T mutant exhibited a 4.8-fold increase in basal production of cAMP, indicating that the receptor was constitutively active. In addition, cells expressing the L469R and D590Y mutants exhibited a 19.1- and 7.8-fold increase in the amounts of basal cAMP, respectively, as compared to those of cells expressing the WT eel LHR (Table 2).
Values are the means±SEM of triplicate experiments. EC50 values were determined from the concentration-response curves obtained in vitro bioassays.
cAMP, cyclic adenosine monophosphate; LHR, luteinizing hormone receptor.
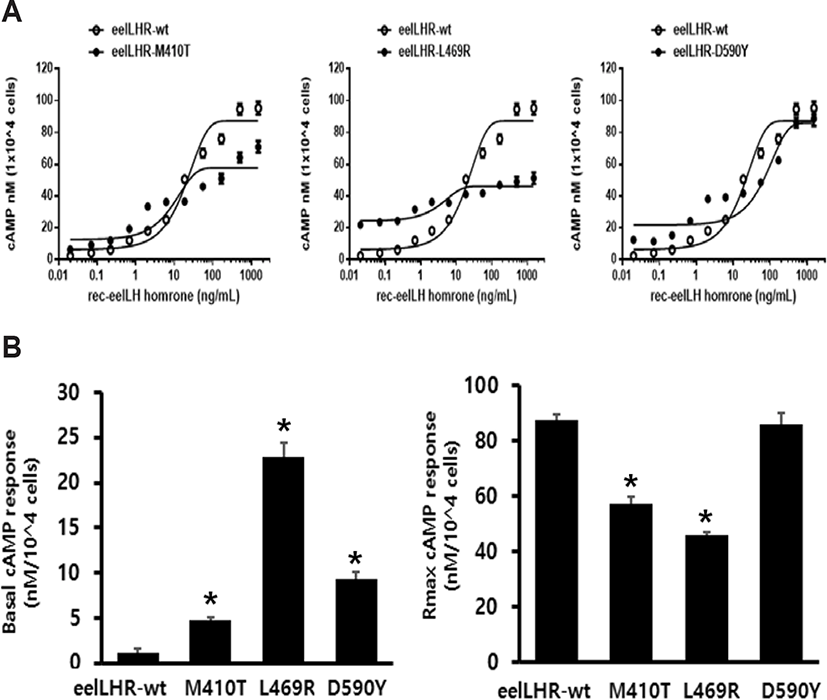
We noted that eel LH produced a concentration-dependent increase in the production of cAMP in cells expressing the activating mutants (M410T, L469R, and D590Y), with an EC50 (50% effective concentration) of 8.7 ng/mL, 3.8 ng/mL, and 77.3 ng/mL, respectively. The maximum cAMP response induced by eel LH in the activating mutants (M410T, L469R, and D590Y) with respect to the maximal response of the WT was found to be approximately 0.65-, 0.52-, and 0.98- fold, respectively. Cells expressing the M410T and L469R mutants were shown to not respond to further stimulation by higher concentrations of agonist. However, the D590Y mutant was observed to reach approximately 98% of the maximal response of the WT.
The concentration-response curve of the accumulation of cAMP in eel LHR-D590Y was characterized by a 4-fold increase in the EC50, but no change in the maximal response when compared with cells expressing WT receptor. Thus, compared with the WT eel LHR, the 3 activating mutations were demonstrated to produce a higher basal cAMP response in CHO-K1 cells, consistent with constitutive activation of the receptor. The mutant exhibiting the highest level of basal production of cAMP (L469R) was found to not react to eel LH with a further increase in maximal cAMP responsiveness. The basal production of cAMP in the L469R mutant represented 49% of the maximal stimulation produced by eel LH (Table 2). Thus, the high basal production of cAMP in the L469R mutant prevented a further increase in the cAMP responsiveness despite a higher agonist stimulation.
To directly assess the functional effects of the 2 inactivating mutations, we transiently expressed these mutant receptors in CHO-K1 cells. The D417N and Y558F mutants were evaluated by quantifying the cAMP stimulation in cells incubated with increasing concentrations of eel LH. As predicted, the cAMP signaling was impaired in cells expressing both mutant receptors compared with those expressing the WT receptor. The basal cAMP response was shown to not be affected by the inactivating mutations, and showed little increase under high concentration of eel LH.
The EC50 value exhibited an approximately 2.6-fold decrease in D417N mutant compared with that of the WT eel LHR. However, the maximal response of this mutant was demonstrated to only be 33% of the response of the WT eel LHR (Fig. 3A). In the Y558F mutant, the EC50 value was also shown to exhibit a 2.2-fold decrease of the value of the WT eel LHR; however, the maximal response was only 25% of that of the WT (Fig. 3B). As shown in Table 3, the signal responsiveness of the receptors was severely affected by the inactivating mutations. The maximal response of these cells was observed to be 67%–75% lower than the maximal response of cells expressing the WT eel LHR.
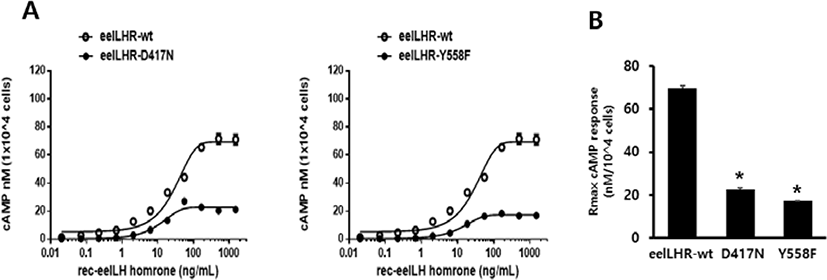
Values are the means±SEM of triplicate experiments. EC50 values were determined from the concentration-response curves obtained in vitro bioassays.
cAMP, cyclic adenosine monophosphate; LHR, luteinizing hormone receptor.
DISCUSSION
The present study was designed to determine the possibility that the activation/inactivation of eel LHRs might be necessary for the signal transduction induced by ligand agonist. In the present study, our results showed that the three activating mutations, eel LHR-M410T, -L469R, and -D590Y, resulted in a distinctly increased basal cAMP response, suggesting that these mutations might cause the constitutive activation of the eel LHR .
Cells expressing the hLHR-M398T mutant (equivalent to M410T in the eel LHR) exhibited high basal levels of cAMP (Yano et al., 1996). Our results also described the M410T constitutively activating mutation in the eel LHR, which was shown to be located in the same second transmembrane region. However, the maximal cAMP response was observed to reach only 65% of the activity detected in cells expressing the WT receptor. The hLHR-L457R mutation (equivalent to L469R in eel LHR) was the first activating mutation identified in hLHR and cells expressing this mutant receptor were noted to exhibit markedly higher basal levels of cAMP (7- to 14-fold) relative to those harboring the WT receptor (Latronico et al., 1998; Galet & Ascoli., 2006; Latronico & Segaloff., 2007; Zhang et al., 2007). Previous results from our colleagues have also shown that basal cAMP responses in cells expressing the rLHR-L435R (equivalent to L469R in eel LHR) displayed a 47-fold increase in the absence of agonist, without leading a further increase in the cAMP response following stimulation by hCG (Min et al., 1998). Previous studies also reported that hLHR-L457R and hFSHR-L460R mutants resulted in strong constitutive activation (Tao et al., 2000; Zhang et al., 2007). However, hFSHR constitutively activating mutants were not as active as hLHR mutants containing the comparable mutation. These results were consistent with our current data, showing a remarkable increase (19.1-fold) in the basal cAMP response of cells expressing the eel LHR-L469R mutant. However, the maximal cAMP response to agonist was shown to be approximately 52% of the WT receptor, as previously described in hLHR (Dhanwada et al., 1996; Huhtaniemi 2000; Huhtaniemi & Themmen, 2005). Hence, we suggested that the L469R mutant did not increase its maximal cAMP responsiveness despite treatment with a high concentration of agonist. Thus, the activating eel LHR-L457R mutant could serve as a specific model in figuring out the basic molecular mechanisms of the activation of eel LHR induced by treatment with high concentration of agonist.
Several years ago, the hLHR-D578Y and -D578G mutants (equivalent to D590Y in the eel LHR) were first reported to be inherited in an autosomal dominant manner, indicating that D578G mutant displayed high basal cAMP response (Shenker et al., 1993; Shenker, 2002). Previous our studies have also reported that rLHR-D556Y (equivalent to D590Y in eel LHR) resulted in a high increase in the concentration of basal cAMP responses with the maximal cAMP response to hCG being about 75% of that of the WT receptor (Min et al., 1998). These results on the hLHR-D578Y and rLHR-D556Y were consistent with our results, indicating that eel LHR- D590Y displayed a high basal cAMP response, and exhibited a maximal cAMP response similar to that of the WT receptor. Thus, our results suggested that the D590Y mutant has a specific characteristic compared with the M410T and L469R mutants, which were markedly decreased in their maximal cAMP activity. This suggested that activating mutations, including eel LHR-M410T,-L469R, and -D590Y might not be unique for the species, and that the basal cAMP response would be markedly increased irrespective of whether these mutants are expressed in mammals (human and rat) or fish.
In the case of the inactivating mutants, we mutated the highly conserved amino acids present in the second (eel LHR-D417) and fifth (eel LHR-Y558) transmembrane helices to asparagine and phenylalanine, respectively. As predicted from results obtained with hLHR and rLHR (Quintana et al., 1993; Min et al., 1998; Tao, 2006), these mutations (eel LHR-D417N and eel LHR-Y558F) were expected to impair signal transduction. Previous studies reported that cells expressing rLHR- D383N displayed a rightward shift in the EC50 for cAMP stimulation, but normal maximal levels (Dhanwada et al., 1996). These results were consistent with our data, showing that eel LHR- D417N and -Y546F were signaling-impairing mutations. However, the maximal response was found to be a little different from our results, displaying only 25%–33% of the maximal response of WT eel LHR. In the present study, the eel LH-induced increase of the production of cAMP in cells harboring the eel LHR-D417N and -Y558F mutations did not completely impair signal transduction. These inactivating mutants were assumed to impair the signal transduction through conformational changes following the formation of the receptor-ligand complexes despite the prolonged treatment with the agonist. These results suggested that the distinct configurations of these mutants induced different signal transduction pathways, resulting in different maximal cAMP responses to LH. The results presented here showed for the first time that three constitutively activating mutants of eel LHR were able to conclusively effect the basal cAMP response.
In conclusion, this study shows that constitutively activating mutations in eel LHR (M410T, L469R, and D590Y) resulted in a significant increase in the basal cAMP production, but cAMP production did not respond to eel LH stimulation with a concentration-dependent increase in M410T and L469R. In contrast, the inactivating mutations (D417N and Y558F) demonstrated to not completely impair the signal transduction. Thus, we suggested that the activation process might involve an agonist-induced conformational change in the receptor. The fundamental mechanisms through which the constitutively activating mutants resulted in a significant increase in the basal cAMP response, whereas the inactivation mutants that impaired signal transduction require further investigation. Future studies using these glycoprotein hormone receptors could provide very valuable information regarding the structure-function relationship of LHR-LH complexes in signal transduction.