




INTRODUCTION
Embryonic stem (ES) cells derived from the inner cell mass (ICM) of embryos have pluripotency and the capacity to differentiate into multiple cell lineages in vitro (Thomson et al., 1998; Reubinoff et al., 2000). Human ES cells have the ability to proliferate in vitro on mouse embryonic fibroblast (MEF) cells and to differentiate into multiple tissue lineages under certain conditions. Human ES cells thus could potentially provide an unlimited source material for future cell therapy, drug screening, and toxicology studies (Ben-Hur et al., 2004; Sartipy et al., 2007). These applications require large amounts of differentiated cells derived from human ES cells. However, human ES cells are difficult to culture and maintain at a large scale. Human ES cells traditionally have been subcultured by manual dissection with a Pasteur pipet and transfer of cell clumps (Oh et al., 2005). This labor-intensive and time-consuming process is not, however, suitable for large-scale production of undifferentiated human ES cells. As an easier alternative method, clumps can be obtained by treatment with collagenase followed by pipetting. The clumps of human ES cells can then be cultured on a feeder layer (Amit et al., 2003) or in a feeder-free condition (Carpenter et al., 2004; Shofren-Janson et al., 2005; Braam et al., 2008; Rosler et al., 2004). A research group proposed an automated method for human ES cell passaging (Joannides et al., 2006). However, it was not confirmed whether this system can maintain the pluripotency of human ES cell cultures for a long period. Future potential clinical applications will require new methods for stable large-scale expansion of human ES cells. Recent studies demonstrated that human ES cells (H9) on feeder cells can be passaged as single cells using accutase (Bajpai et al., 2008). Furthermore, maintenance of human ES cells (SA001, SA002, SA002.5, SA121 and SA167) passaged as single cells using Trypsin-EDTA and TrypLE on a feeder layer was also reported (Ellerstrom et al., 2007).
Karyotypic changes present a critical pitfall in human ES cell cultures. Normal human ES cells have a stable diploid karyotype (Thomson et al., 1998) and the chromosomal integrity of human ES cells is believed to persist after prolonged in vitro culture. In particular, ES cells passaged by manual dissection of colonies are known to retain a stable karyotype (Buzzard et al., 2004). However, several groups independently showed chromosomal changes in human ES cell lines. The Draper group observed karyotypic change in human ES cell lines H1, H7, and H14. Human ES cells maintained for several months by the conventional passaging approach retained an undifferentiated phenotype and normal karyotype, but long-term culture (about 25 passages) of defrosted cells after freezing showed mutation in chromosome 17. Human ES cells passaged under a feeder-free condition (~25 passages) also showed mutation in chromosome 17 and chromosome 12 (Draper et al., 2004). Furthermore, change of in vitro culture conditions such as recycling of the feeder increased the survival and growth of ES cells and also maintained expression of human ES cell markers; however it induced deleted mutation on chromosome 7 and trisomic mutation on chromosome 12 in HS181 human ES cells (Imreh et al., 2006). Another group (Caisander et al., 2006) reported trisomic mutation in chromosome 13 in a variety of human ES cell lines (SA002, SA002.5, SA121, and SA167) after 19 passages. Furthermore, passage of cell clumps obtained by an enzymatic method also appears to compromise the genetic mutation of human ES cells on the feeder layer or in a feeder-free condition (Maitra et al., 2005; Mitalipova et al., 2005). Recently, the International Stem Cell Initiative analyzed the genomic stability during the culture of 125 human ES cell lines. As ES cells cultured by most groups retained the normal karyotype, it is likely that chromosomal instability is not an intrinsic property of human ES cells but appears to derive from improper culture conditions (International Stem Cell Initiative, 2011).
In this study, we observed that H9 human ES cells can be passaged and cultured in a feeder-free condition after single cell dissociation using accutase, maintaining stem cell marker expression and normal karyotype. However, we found frequent chromosomal modification when we made frozen stocks and cultured defrosted ES cells as single cells.
MATERIALS AND METHODS
H9 human ES cells (WiCell Research Institute, Madison, USA) were maintained on MEF cells in an ES medium containing 80% DMEM/F12 (Gibco, Carlasbad, USA), 20% KnockOut serum replacement medium (Gibco), 1 mM L-glutamine (Sigma-Aldrich, St. Louis, USA), 0.1 mM β-mercaptoethanol (Sigma-Aldrich), 1% nonessential amino acids (Gibco), and 4 ng/ml human basic fibroblast growth factor (bFGF). Human ES cells were detached from the plates by incubation in a 2.5 mg/ml collagenase IV (Gibco) solution for 10-20 min at 37°C. Human ES colonies were washed with DMEM/F12 medium twice and then physically dissociated to small clumps. Human ES cell clumps were moved into the MEF plated dish. The culture medium was changed daily.
Human ES cell clumps were detached from the plates by incubation in a 2.5 mg/ml collagenase IV solution for ~20 min at 37°C. ES cell clumps were washed with DMEM/F12 medium and then incubated at 37°C for 5 min with accutase (Chemicon, Temecula, USA) and TrypLE (Gibco), respectively. After complete dissociation into single cells, human ES cells were collected by centrifugation (1,200 rpm for 3 min) and washed with DMEM/F12. Dissociated human ES cells were replated at a density of 1×105 cells/ml on plates coated with Matrigel (BD Biosciences, San Jose, USA) and cultured in a feeder-free medium containing 70% MEF conditioned medium, 30% ES medium, and 10 ng/ml human bFGF. The human ES cells were passaged between days 5-7. The ES cell medium was changed daily.
Cells were fixed and permeabilized using cytofix/cytoperm™ (BD Biosciences). The cytofix/cytoperm™wash solution was added to the cells together with individual primary antibodies and incubated for 1h. The antibodies used in this study are as followings. Anti-Oct-3/4 (BD Biosciences); anti-SSEA-1 and anti-SSEA-4 (Developmental Studies Hybridoma Bank, Iowa City, USA); anti-TRA-1-60 and anti-TRA-1-81 (Chemicon), and anti-Sox2, anti-Klf-4, and anti-Nanog (Santa Cruz Biotechnology, Inc., Santa Cruz, USA). After washing with cytofix/cytoperm™ wash solution, the cells were incubated with the FITC-conjugated secondary antibody in cytofix/cytoperm™ wash solution at 4°C in the dark for 1h. After washing, samples were analyzed with FACScan (BD Biosciences).
Total RNA was isolated using TRI Reagent® according to the instructions provided by the manufacturer (MRC, Cincinnati, USA). 5 μg of total RNA was reverse-transcribed in the first-strand buffer containing 6 μg/ml oligo(dT) primer, 50 U StrataScript™ reverse transcriptase (Invitrogen, Carlsbad, USA), 2 mM dNTP, and 40 U RNase block ribonuclease inhibitor. The reaction was conducted at 42°C for 1 h. One microliter of the cDNA synthesis was subjected to the standard PCR reaction for 30 cycles of denaturation for 60 sec at 95°C, annealing for 60 sec at 58°C, and elongation for 60 sec at 72°C. The primer sequences used in this study are as follows. GAPDH, 5’-ACCACAGTCCATGCCATCAC-3’ (sense) and 5’-TCCACCACCCT GTTGCTGTA-3’ (anti-sense) (product size 452 bp). OCT4, 5’-GGAAAGGCTTCCC CCTCAGG-3’ (sense) and 5’-AAGAACATGTGTAAG CTGCGG-3’ (antisense) (313 bp). Nanog, 5’-GGAAG ACAAGGTCCCAGTCA-3’ (sense) and 5’-CTGGTTGC TCCAGGTTGAAT-3’ (antisense) (364 bp).
Karyotype of the human ES cells was analyzed by G-banding by Trypsin using Giemsa (GTG-banding) (Seol et al., 2007). Human ES cells were harvested and fixed. The human ES cells at metaphases were karyotyped using a chromosome imaging analyzer system (GenDix, Seoul, Korea).
RESULTS AND DISCUSSION
Maintenance of human ES cells with pluripotency requires labor-intensive and time-consuming work to subculture cells. Previously, other groups have shown that human ES cells can be maintained on MEF cells after single cell dissociation with enzymes such as collagenase and accutase. They confirmed efficient propagation of accutase-dissociated human ES cells (Bajpai et al., 2008). Accutase-passaged human ES cells exhibited a higher proportion of undifferentiated human ES cells as compared to collagenase-passaged cells (Bajpai et al., 2008). In contrast, cells dissociated with trypsin revealed much lower growth. Here, we dissociated H9 human ES cells to single cells with accutase and successfully expanded the cells in a feeder-free condition.
We cultured human ES cell line H9 in a feeder-free condition on Matrigel-coated plates after single cell dissociation using accutase until 39 passages. Prolonged culture of ES cells using the enzyme accutase may adversely affect the stem cell properties of human ES cells. Therefore, we first examined expression of ES cell markers during the culture period (Fig. 1). We performed a FACS analysis for stem cell marker expression in ES cells at early passage (passage 1, Fig. 1A) and at a later passage (passage 26, Fig. 1B). ES cell markers such as SSEA-4, Oct4, Nanog, Tra-1-60, and Tra-1-81 were ordinarily expressed in both cultures. Expression of embryonic stem cell markers at the mRNA level was also consistently observed by a RT-PCR analysis (Fig. 1C): expression levels of Oct4 and Nanog were similar in cells at early passage and late passage (passage 1 and passage 39, respectively). These results suggest that the consecutive passaging of human ES cells after single cell dissociation with accutase permits maintenance of an undifferentiated state of ES cells, at least in the context of ES cell marker expression.
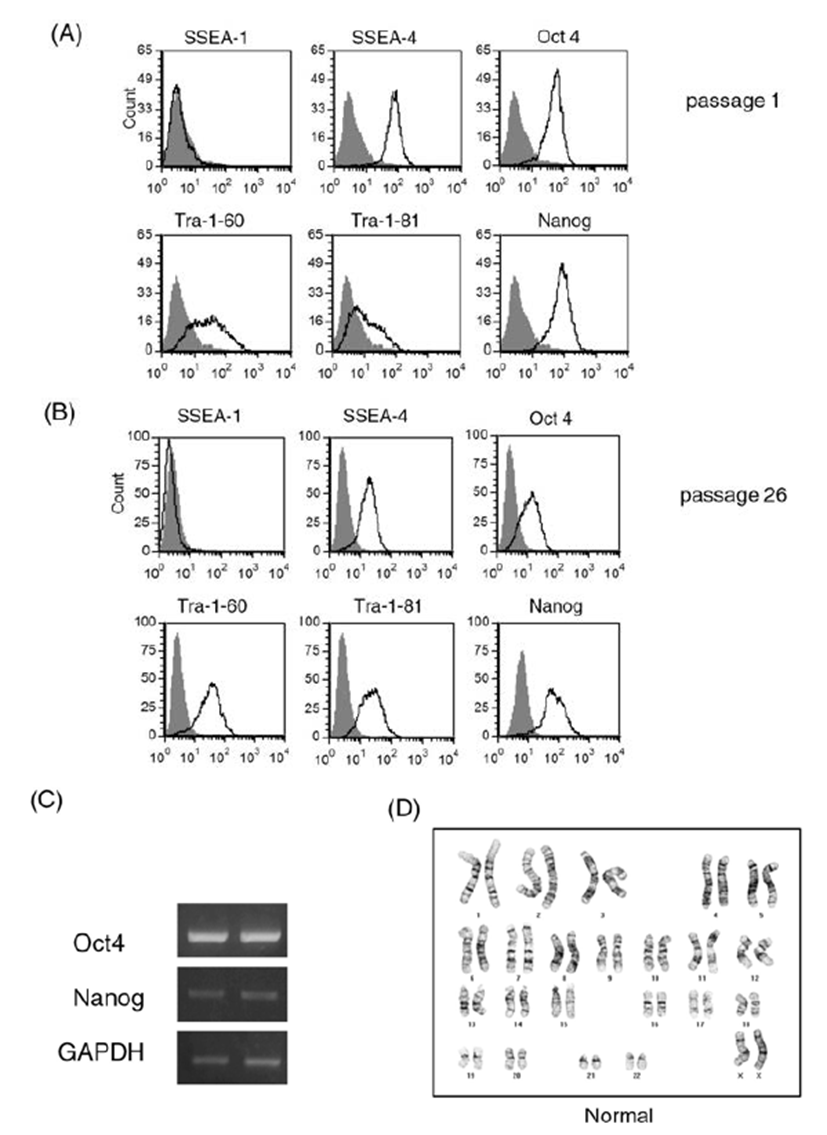
Long-term enzymatic passaging is reported to induce karyotypic abnormalities in human ES cells (Buzzard et al., 2004; Mitalipova et al., 2005). To confirm the karyotypes of human ES cells, we analyzed the chromosomal status of ES cells using the GTG-banding method and monitored whether translocations and/or aneuploidies are found (Fig. 1D). After 20 enzymatic passages using accutase, the human ES cells had normal karyotype. From these results, we confirmed that the human ES cell H9 can be efficiently expanded in a feeder-free condition after single cell dissociation using accutase.
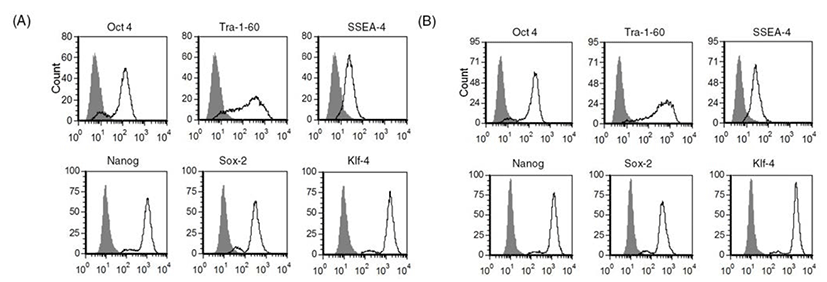
As we confirmed that human ES cells can be maintained by culture of single cells dissociated with accutase, we next investigated whether other enzymes such as TrypLE can be used in the same way to culture human ES cells as single cells. As shown in Fig. 2, expression of stem cell markers in TrypLE-passaged human ES cells was analyzed by FACS. Expression levels of stem cell markers such as Tra-1-60, SSEA-4, Nanog, Sox-2, and Klf-4 were similar in accutase-passaged cells (Fig. 2A) and TrypLE-passaged cells (Fig. 2B). These results reveal that TrypLE may be used as an alternative to accutase in the culture of human ES cell H9 after single cell dissociation.
To routinely culture human ES cells, early passage cells should be stored as frozen stocks and should be thawed and expanded. Therefore, we froze accutasepassaged human ES cells at several passages and analyzed properties of the accutase-passaged human ES cells after freeze-thawing (Fig. 3).
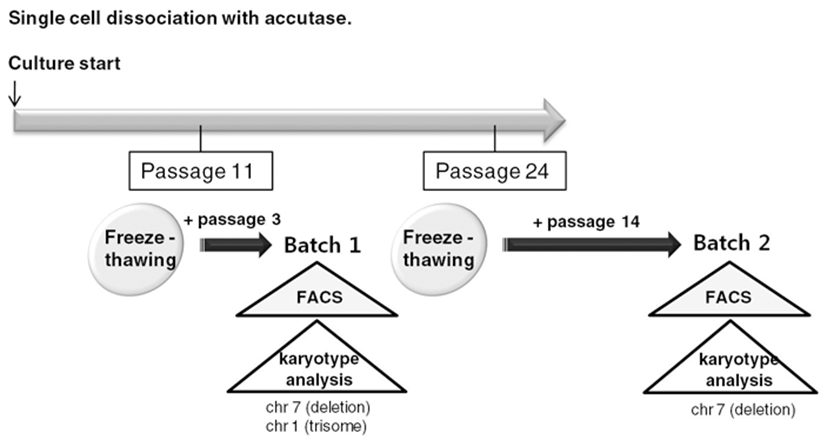
We selected two of the stocks (frozen at passage 11 and passage 24, respectively) and cultured them on Matrigel-coated plates after usual thawing. We cultured the stock frozen at passage 11 for another 3 passages and performed a FACS analysis to assess expression of ES cell markers (Batch 1) (Fig. 4A). We cultured the other stock (passage 24) for another 14 passages and checked ES cell marker expression (Batch 2) (Fig. 4B). Both of the human ES cell batches expressed ES cell markers, such as Tra-1-60, Tra-1-81, Sox2, Klf-4, Oct4, and Nanog. Expression levels of ES cell markers were similar in both cultures. These results suggest that accutasepassaged human ES cells may maintain an undifferentiated state after single cell passaging even after freeze-thawing.
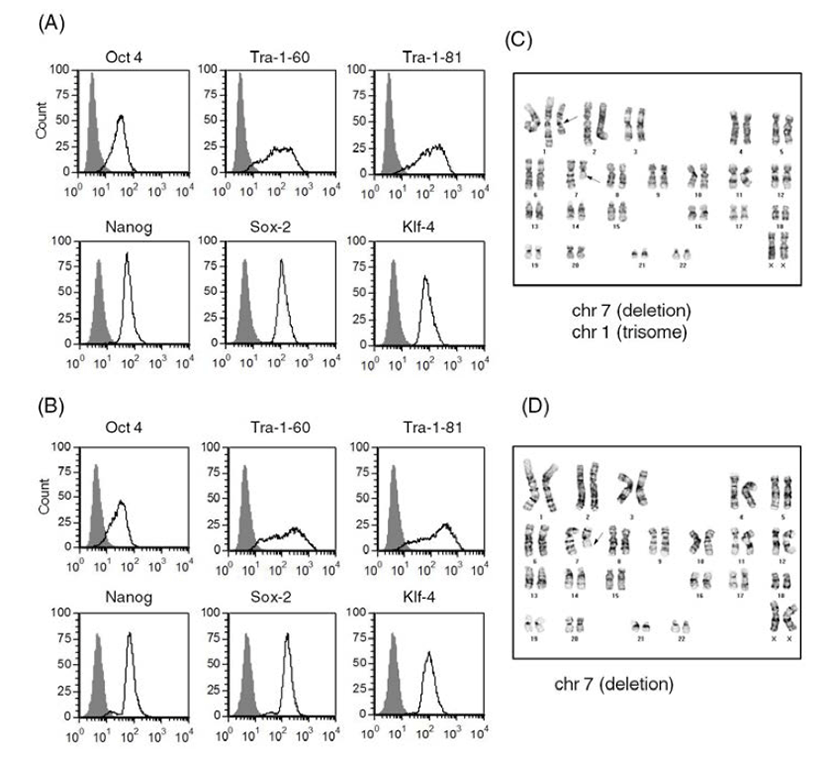
Next, we checked the karyotype of the human ES cells cultured after freeze-thawing. As shown in Fig. 4C and Fig. 4D, both of the cultures (Batches 1 and 2) had abnormal karyotypes; one culture (batch 1) had a deletion mutation in chromosome 7 and trisomy of chromosome 1 (Fig. 4C) and the other culture (batch 2) had a deletion mutation in chromosome 7 (Fig. 4D). Considering that the accutase-passaged cells showed normal karyotype, it is likely that the karyotypic abnormalities derived from the culturing process after thawing. When we culture ES cells after freeze-thawing, the survival rate is very low, even with usual cell clumps. Previously, another group reported that human ES cell lines with high replating efficiency selected from an ES cell culture after single cell dissociation with trypsin had deletion mutations or trisomic mutations in chromosomes 1, 7, 12 or 17 (Hasegawa et al., 2006; Gertow et al., 2007). Therefore, it is possible that a subpopulation of genetically unusual human ES cells were selected and propagated during the culture period, especially after freeze-thawing of the accutase-passaged ES cells. This kind of selection has also been reported in human ES cells under bulk culture conditions (Buzzard et al., 2004; Mitalipova et al., 2005; Imreh et al., 2006). Considering the genetic abnormality found in the ES cells of batch 1, which were cultured for only 3 passages after freeze-thawing, storing accutase-passaged cell as a frozen stock is not recommended: consecutive culture of accutase-passaged human ES cells after freeze-thawing may induce abnormal karyotypes with high risk. Even though we did not examine the karyotype of TrypLE-passaged human ES cells after freeze-thawing, we expect that similar results will be obtained. Previously, it was reported that a ROCK inhibitor Y-27632 improves survival of cryopreserved serum/feeder-free single human ES cells (Li et al., 2009). Furthermore, extended incubation with feeder cells overnight before vitrification was reported to enhance the survival and maintenance of undifferentiated state of cryopreserved human ES cells (Cha et al., 2008). Therefore, application of this information in our culture system can be a strategy to reduce the risk of genetic instability.
From the obtained results, we confirmed that the culture method of single cell-dissociated human ES cells is useful for obtaining a large number of undifferentiated cells. We also identified a critical pitfall of the culture method: extreme genetic instability of human ES cells during the culture after freeze-thawing. Our information on the characteristics of ES cells during human ES cell culture through single cell dissociation will contribute to the establishment of more efficient culture methods and better usage of human ES cells.