
INTRODUCTION
In vertebrate embryonic head, a series of pouches arises in the pharyngeal endoderm. The pharyngeal pouches are important for craniofacial development. First, they are required for facial skeletons by segmenting the pharyngeal arches and providing survival signals, such as sonic Hedgehog, to the arches (Piotrowski & Nüsslein-Volhard, 2000; Brito et al., 2006). Then, they further differentiate into endocrine glands in the head and neck, including the thymus and parathyroid (Grevellec & Tucker, 2010). In mice and zebrafish, abnormal development of pouches resulted in loss or malformation of facial cartilages and hypoplasia of the thymus and parathyroid (Piotrowski & Nüsslein-Volhard, 2000; Dickmeis et al., 2001; Kikuchi et al., 2001; Grevellec & Tucker, 2010). In humans, defects in pouches are associated with DiGeorge syndrome, a congenital craniofacial disability, with TBX1 being identified as a key gene in the etiology of the syndrome (Piotrowski & Nüsslein-Volhard, 2000; Lindsay et al., 2001; Tran et al., 2011). Recently, the genetic and cellular basis underlying pouch formation has been revealed in vertebrates. Loss of Tbx1, Pax1, and Foxi1 transcription factors and Fgf ligands led to loss or malformation of pouches and facial skeletons in mice and zebrafish (Lindsay et al., 2001; Nissen et al., 2003; Piotrowski et al., 2003; Tran et al., 2011; Okada et al., 2016; Jin et al., 2018; Liu et al., 2020). In zebrafish, Wnt, Ephrin, and VEGF-C signaling molecules are also required for pouch formation, with Neuropilin signaling being implied in pouch formation (Crump et al., 2004; Ober et al., 2004; Choe et al., 2013; Choe & Crump, 2014; Choe, 2023). To control pouch formation, Pax1 regulates the expression of tbx1 and fgf3 in the pharyngeal endoderm in medaka and zebrafish, with Foxi1 regulating wnt4a expression in zebrafish (Okada et al., 2016; Jin et al., 2018; Liu et al., 2020). At the cellular level, Tbx1 promotes a collective migration of pouch-forming cells through Wnt11r and Fgf8a, and then the migrating pouch-forming cells are matured into a bi-layered pouch by Wnt4a and Ephrin B2 and B3 ligands in zebrafish (Choe et al., 2013; Choe & Crump, 2014). Besides the roles of these transcription factors and signaling molecules, a lot of genetic and cellular mechanisms of pouch formation have not been uncovered yet. To better understand the genetic basis of pouch formation, here we analyzed the role of Fgf signaling in developing pouches in zebrafish.
In mice, Fgf signaling is implicated in pouch formation. Loss of Fgf3, Fgf8, Fgf receptor (Fgfr) 1, or 2 resulted in abnormal pouches (Aggarwal et al., 2006; Jackson et al., 2014). For pouch formation, Fgf3 and Fgf8 were coexpressed in different combinations with Tbx1 and were strongly downregulated in Tbx1 mutant embryos (Aggarwal et al., 2006). Interestingly, pharyngeal endoderm-specific depletion of Fgfr1 and Fgfr2 displayed more severe pouch defects than mutants for Fgf3 or Fgf8 did, implying that additional Fgf genes might be involved in pouch formation (Jackson et al., 2014). Furthermore, epistatic analysis of Tbx1, Fgf3, and Fgf8 genes suggested potential functional redundancy with additional Fgfs in the development of the pharyngeal pouches via Tbx1 (Aggarwal et al., 2006). Indeed, Fgf4 and Fgf16 are also expressed in pharyngeal endoderm, with their roles in pouch formation not being determined (Niswander & Martin, 1992; Wright et al., 2003). In zebrafish, redundant essential functions of fgf3 and fgf8a are also implicated in pouch formation; knockdown of fgf3 in fgf8a mutants caused more severe defects in pouch formation than fgf8a mutants did (Crump et al., 2004). Also, it was suggested that fgfr1a, fgfr1b, and fgfr2 acted redundantly to regulate pouch development (Leerberg et al., 2019). A high degree of redundancy in Fgf signaling implicated in pouch formation suggests additional Fgf genes in the development of the pharyngeal apparatus in mice and zebrafish. However, besides fgf3 and fgf8a, the requirement for other fgf genes in pouch formation has not yet been analyzed in zebrafish.
In order to identify additional fgf genes acting in pouch formation in zebrafish, we looked at individual fgf genes that are expressed in the pharyngeal endoderm. We found that three fgf genes, fgf4, fgf24, and fgf17, were expressed in distinct regions of the pharyngeal endoderm in the morphogenesis of pouches. Furthermore, we observed that they acted downstream of Tbx1 or Fgf8a in pouch formation; their expression was strongly downregulated in tbx1 or fgf8a mutant embryos, with their expression being barely affected in fgf3 mutants. However, single, all double mutant combinations, and triple mutant for fgf4, fgf24, and fgf17 showed normal pouches and facial skeletons. Our results suggest that fgf4, fgf24, and fgf17 could be involved in pouch formation, with a high degree of redundancy.
MATERIALS AND METHODS
Zebrafish were grown according to the Animal Protection Act (2017) in Korea. All zebrafish work was approved by Gyeongsang National University Institutional Animal Care and Use Committee. Published lines include tbx1tu285 (Piotrowski et al., 2003), fgf3t24152 (Herzog et al., 2004), fgf8ati282a (Reifers et al., 1998), fgf24tm127c (van Eeden et al., 1996), Tg(~3.4her5:EGFP) (Tallafuss & Bally-Cuif, 2003), and Tg(nkx2.3:Gal4VP16) (Choe et al., 2013). fgf4 and fgf17 mutant lines were generated with CRISPR/Cas9 system (Hwang et al., 2013). 150 pg of gRNA and 150 pg of mRNA encoding a nuclear-localized Cas9 were injected together into one-cell stage wild-type Tübingen (TU) embryos harboring Tg(~3.4her5:EGFP). Injected embryos were grown and outbred to wild-type TU animals to identify zebrafish bearing in/del mutations in fgf4 and fgf17 genes. Three mutant lines for fgf4 (fgf4GNU6, fgf4GNU8, and fgf4GNU9) and three for fgf17 (fgf17GNU1, fgf17GNU4, and fgf17GNU5) were secured. fgf4GNU8 and fgf17GNU1 were used for this study. For genotyping of fgf4GNU8, a PCR amplified fgf4 fragment with primers fgf4_GT_ F and fgf4_GT_R, was digested with AccI, with a wild-type fragment producing 358 bp and mutant fragment generating 176 and 178 bp. For genotyping of fgf17GNU1, primers fgf17_GT_ F and fgf17_ GT_R converted fgf17GNU1 into a codominant polymorphism, with a wild-type product of 147, 108 and 90 bp and mutant products of 245 and 90 bp after TauI digestion. Tg(UAS:DN-Fgfr1) transgenic construct was generated using the Gateway (Invitrogen, Carlsbad, CA, USA) Tol2kit (Kwan et al., 2007). For pME-DN-Fgfr1, a dominant-negative form of Fgfr1 that is missing its intracellular kinase domain was produced by PCR using primers DN-Fgfr1-B1F and DN-Fgfr1-B2R from multi-stage zebrafish cDNA (Fürthauer et al., 2004). 30 ng of UAS:DN-Fgfr1 transgenic construct and 35 ng of DNA construct bearing Tol2 transposase gene were injected into one-cell stage wild-type TU embryos. Five independent transgenic lines for Tg(UAS:DN-Fgfr1:pA) were established based on α-crystallin:Cerulean eye fluorescence. Primers are listed in Table 1.
Immunohistochemistry for Alcama (Zebrafish International Resource Center, Eugene, OR, USA, 1:400), fluorescent in situ hybridization in conjunction with GFP immunohistochemistry (Torrey Pines Biolabs, 1:1,000), and Alcian blue and alizarin red staining were performed as described previously (Crump et al., 2004; Zuniga et al., 2011). Partial cDNA fragments of fgf4, fgf24, and fgf17 were amplified from mixed-stage zebrafish cDNA. The PCR fragments were cloned into the pGEM®-T easy vector (Promega, Madison, WI, USA), and Digoxigenin (DIG)-labeled antisense riboprobes were transcribed with T7/SP6 RNA polymerase (Roche Life Sciences, Basel, Switzerland) from linearized plasmids. Primers used for riboprobes are listed in Table 2.
| Gene | Forward primer (5’ to 3’) | Reverse primer (5’ to 3’) |
|---|---|---|
| fgf4 | TTGCCAATCCTGGTCTTA | GAAAGTGCATCGTCGTTC |
| fgf24 | TTATATCGCGTGCTGGAT | CCACAAGCAGAGCGTACT |
| fgf17 | GAGAACGCCAGACATGAG | AGGCGTTGAAGATGAACA |
Fluorescent images for immunohistochemistry and in situ hybridization were captured with an Olympus FLUOVIEW FV3000 confocal microscope using FV31S-SW software (Olympus, Tokyo, Japan). We took approximately 100 μm Z-stacks at 3.5-μm intervals with an Olympus UPLXAPO 20× objective lens and projected them into a single image. Flat-mounted facial cartilages and bones dissected manually from Alcian Blue and alizarin red staining larvae were imaged with an Olympus BX50 upright microscope using mosaic V2.1 software (Tucsen Photonics, Fuzhou, China). Any adjustments were applied to all panels using Adobe Photoshop (Adobe Systems, San Jose, CA, USA).
RESULTS
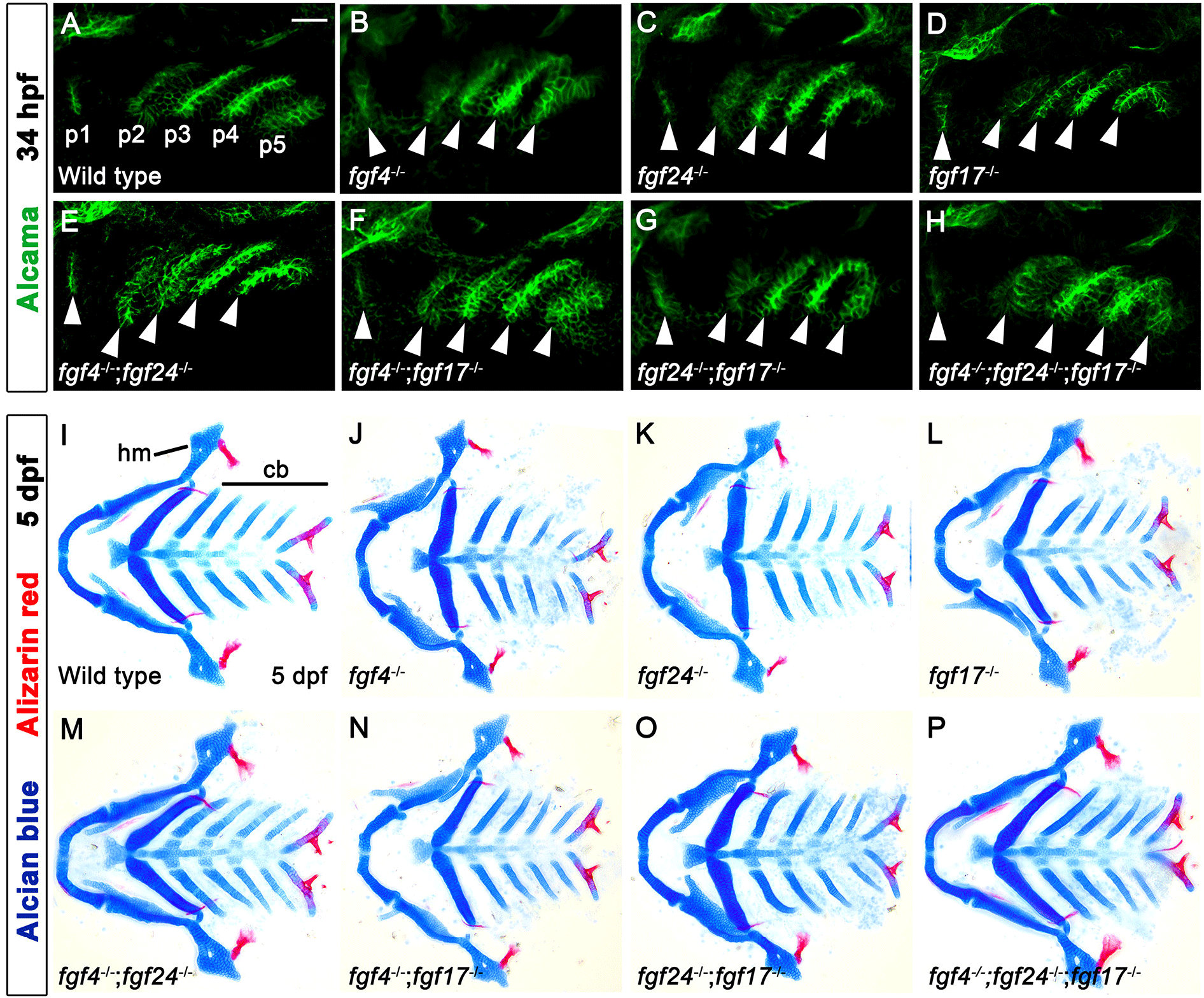
In wild type, a total of six pouches form at 34 hours post-fertilization (hpf), with the sixth pouch hardly seen (Fig. 1A). Although fgf3 and fgf8a are essential for pouch formation, the loss of fgf3 and fgf8a resulted in mild defects in pouches in that the anterior two pouches were less affected, with the posterior four pouches being malformed or absent (Fig. 1B and C). Consistently, the development of hyomandibular (hm) cartilage that is controlled by the first pouch was less affected, whereas that of ceratobranchial (cb) cartilage that is regulated by the posterior pouches was severely defective in single mutant for fgf3 and fgf8a at 5 days post-fertilization (dpf) (Fig. 1E–G). If Fgf signaling is critical for the development of all six pouches, the absence of Fgf signaling in the pharyngeal endoderm would be able to cause defects in all pouches, and hm and cb cartilages whose development relied on all six pouches (Crump et al., 2004). We utilized a dominant-negative approach to test this hypothesis. To do so, we generated Tg(UAS:DN-Fgfr1a) lines harboring a dominant-negative mutant allele of fgfr1a. Utilizing the pharyngeal endoderm driver Tg(nkx2.3:Gal4VP16) lines that are able to express target genes in pouch-forming endoderm, including the anterior pouches (Choe et al., 2013), we expressed DN-Fgfr1a specifically in the endoderm during pouch formation. Pharyngeal endoderm-specific expression of the DN-Fgfr1a blocking Fgf signaling in the endoderm resulted in the absence of all pouches at 34 hpf as well as defects in hm and cb cartilages at 5 dpf (Fig. 1D and H). This result suggests that Fgf signaling, which is required in the endoderm, is essential for the formation of all pouches.
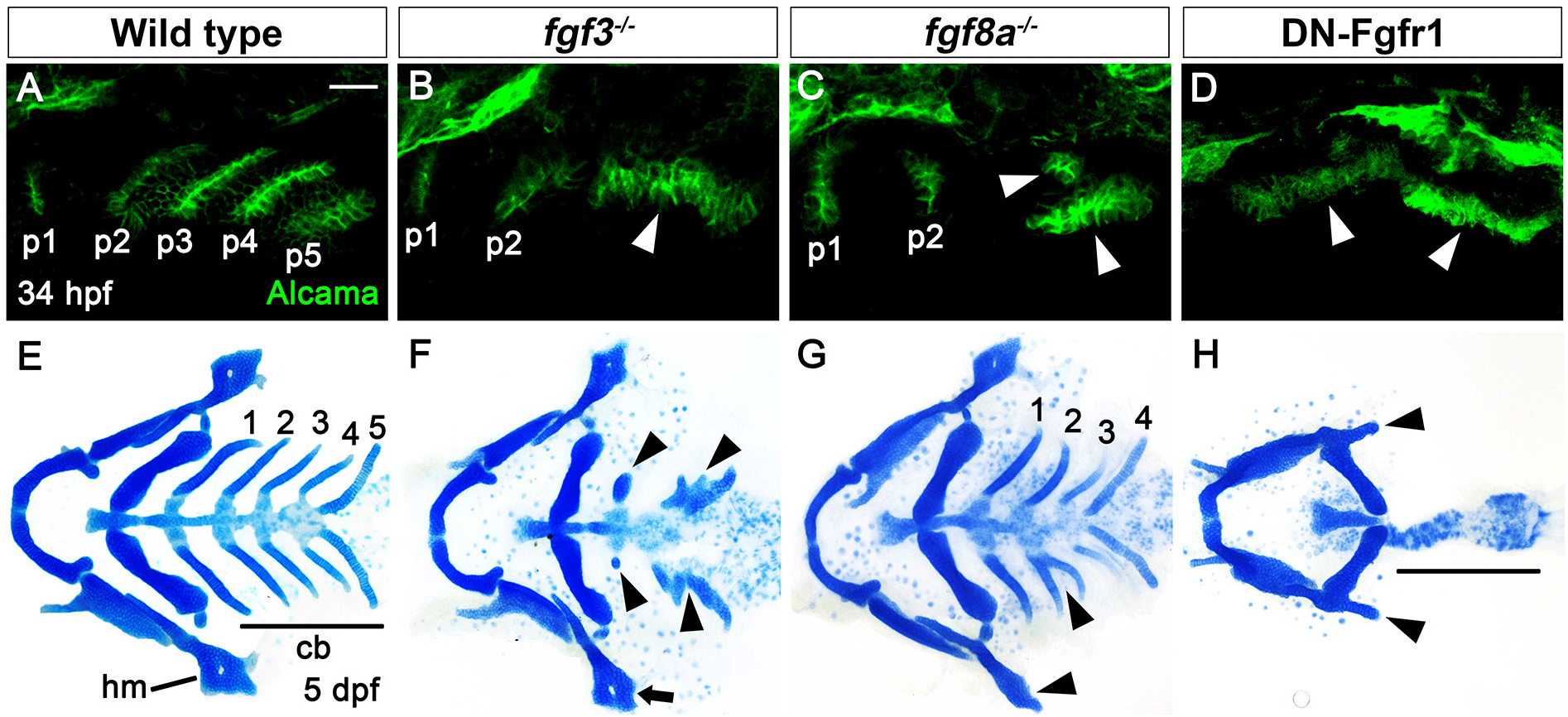
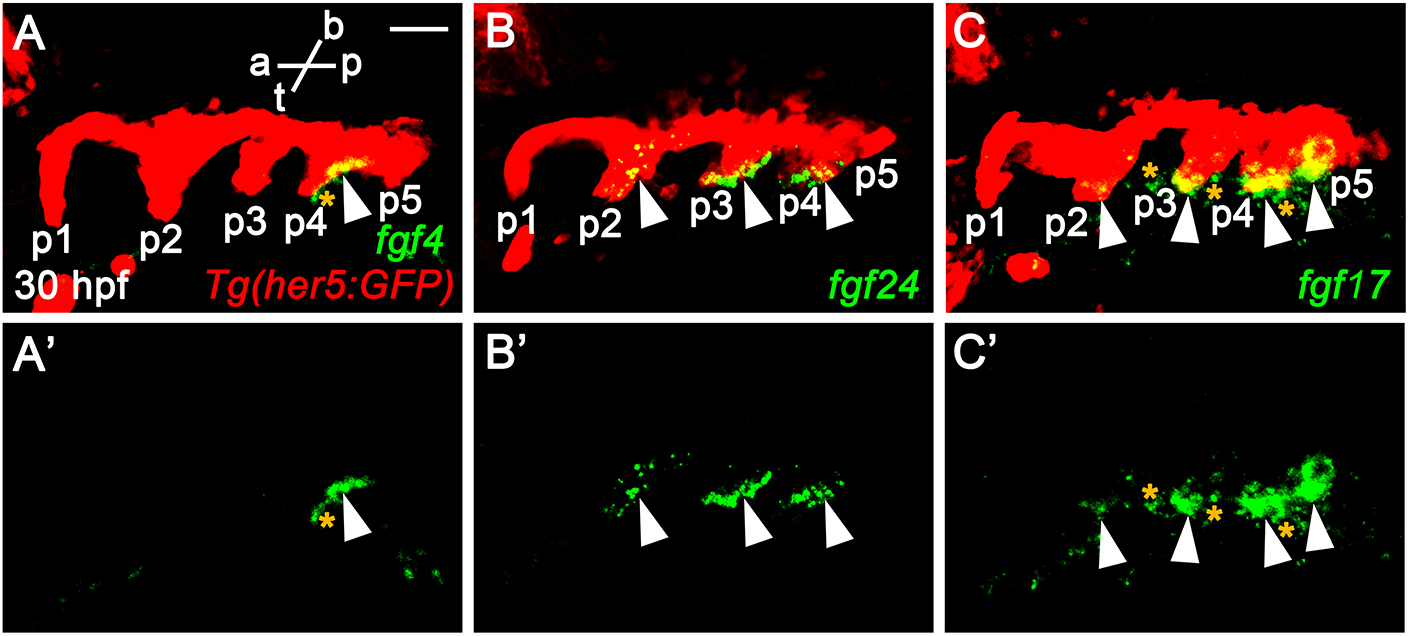
The phenotypic difference in pouches among fgf3 mutant, fgf8a mutant, and endoderm-specific block of Fgf signaling suggests that additional fgf genes might be necessary for the development of pouches and associated facial cartilages, in addition to fgf3 and fgf8a. To identify additional fgf genes necessary for pouch formation, we first examined the expression of fgf genes in the pharyngeal endoderm at 30 hpf, in which four pouches form and the fifth pouch is just being established. To do so, we performed in situ hybridization for candidate fgf genes in wild-type embryos harboring Tg(her5:GFP) transgene, a pharyngeal endoderm and pouches reporter (Tallafuss & Bally-Cuif, 2003). Consequently, we found that fgf4, fgf24, and fgf17 were expressed in the distinct regions of pharyngeal pouches at 30 hpf. fgf4 was expressed locally in the basal region of the fourth pouch, with its expression not seen in the anterior three pouches and the forming fifth pouch (arrowheads in Fig. 2A and A’). It was also expressed in the adjacent mesoderm to the fourth pouch (asterisks in Fig. 2A and A’). Transcripts for fgf24 were observed in the second to fourth pouches, with those in the second pouch being faded compared to those in the third and fourth pouches (arrowheads in Fig. 2B and B’). They were also restricted in the posterior layer of the bi-layered pouches. However, they were not seen in the first and forming fifth pouches (Fig. 2B and B’). Apparently, fgf24 was not expressed in the adjacent mesoderm to the pharyngeal endoderm at 30 hpf. Next, we found that fgf17 was expressed at the tip of the second to fifth pouches, with its expression being faded in the second pouch at 30 hpf (arrowheads in Fig. 2C and C’). At the tip of pouches, fgf17 was expressed in both anterior and posterior layers. It was also expressed in the mesoderm adjacent to the pharyngeal endoderm (asterisks in Fig. 2C and C’). Expression of fgf4, fgf24, and fgf17 in the distinct regions of pharyngeal pouches suggests that they could play a role in pouch formation during craniofacial development.
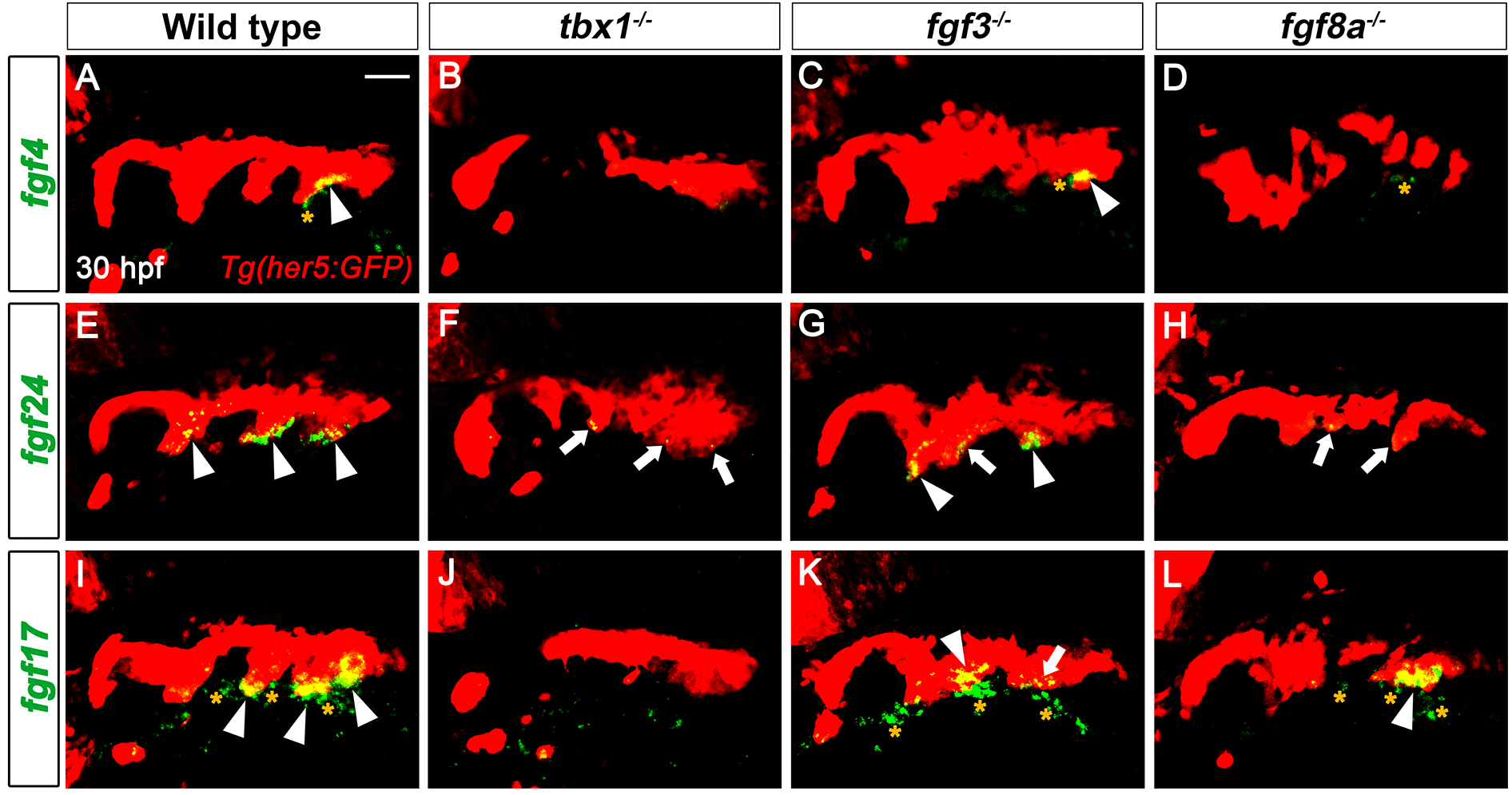
Expression of fgf4, fgf24, and fgf17 in developing pouches at 30 hpf implies their potential roles in pouch formation and facial cartilage development. Since tbx1, fgf3, and fgf8a are key genes controlling pouch formation, we next analyzed whether the three fgf genes acted downstream of the key genes for pouch formation. Compared to the wild type, the endodermal expression of fgf4, fgf24, and fgf17 in the pouches was significantly reduced in tbx1 mutants in which most pouches did not form (Fig. 3A, B, E, F, I, and J). The mesodermal expression of fgf4 and fgf17 also being abolished in tbx1 mutants at 30 hpf (Fig. 3B and J). Since Tbx1 acts in both pharyngeal endoderm and adjacent mesoderm for pouch formation (Choe & Crump, 2014), the endodermal and mesodermal expression of the three fgf genes require Tbx1 activity. In contrast to Tbx1, Fgf3 appears to be dispensable for the expression of the three fgf genes in the pharyngeal regions at 30 hpf. In fgf3 mutants where only the anterior one or two pouches form, fgf4 expression in the endoderm was normal (arrowhead in Fig. 3C). Although the endodermal expression of fgf24 and fgf17 was slightly reduced in some potential pouch regions (arrows in Fig. 3G and K), their expression was fairly normal in the endoderm of fgf3 mutants (arrowheads in Fig. 3G and K). Also, the mesodermal expression of fgf4 and fgf17 was seen in fgf3 mutants at 30 hpf (asterisks in Fig. 3C and K). The regulation of fgf4 and fgf24 by Fgf8a was similar to that by Tbx1 in that the endodermal expression of fgf4 and fgf24 were abolished or significantly reduced in fgf8a mutants (Fig. 3D and H). However, in fgf8a mutants, the endodermal expression of fgf17 was not affected (Fig. 3L). In fgf8a mutants, the mesodermal expression of fgf4 was unaffected, whereas that of fgf17 was reduced (Fig. 3D and L). Thus, in the pharyngeal regions, the genetic regulation of the three fgf genes by Fgf8a is not necessarily identical to that by Tbx1. Misregulation of fgf4, fgf24, and fgf17 in single mutants for tbx1 and fgf8a implies that Tbx1 and Fgf8a could control pouch and facial cartilage formation through these fgf genes during craniofacial development.
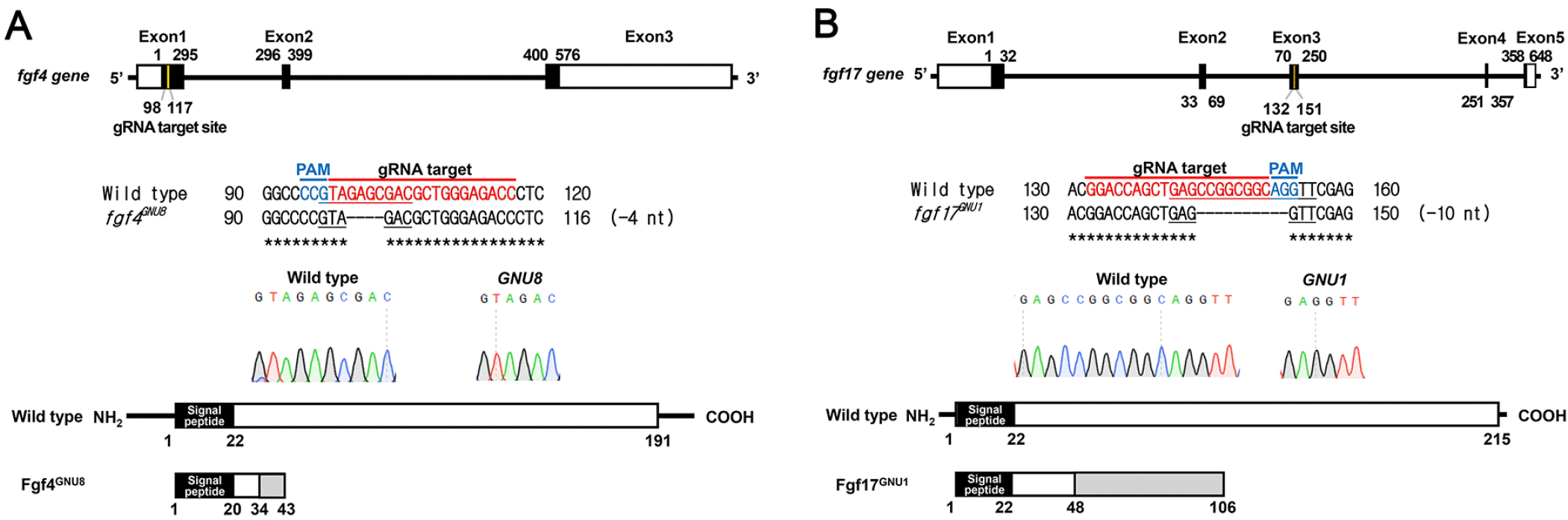
If fgf4, fgf24, and fgf17 are required for the development of pouches and associated facial cartilages as downstream targets of Tbx1 and Fgf8a, loss-of-function mutations in these fgf genes would result in defects in pouches and facial cartilages. In order to access a role for fgf4, fgf24, and fgf17 in the development of pouches, we generated loss-of-function mutations in fgf4 and fgf17 genes with CRISPR/Cas9 system and secured a fgf24 mutant available in the zebrafish community (van Eeden et al., 1996). For fgf4 mutants, we established a four-nucleotide deletion allele in the fgf4 gene (fgf4GNU8), compared to wild-type allele (Fig. 4A). While wild-type allele produces 191 amino acids including 20 amino acids of a signal peptide, the mutant allele was predicted to produce only 34 normal amino acids including 20 amino acids of a signal peptide and an extra 9 amino acids (Fig. 4A). Thus, fgf4GNU8 seems to be a loss-of-function allele. For fgf17 mutants, we secured a ten-nucleotide deletion allele in the fgf17 gene (fgf17GNU1), which was predicted to produce 48 normal amino acids and an extra 58 amino acids that were completely different from those of wild-type Fgf17 (Fig. 4B). Thus, it is likely that fgf17GNU1 is also a loss-of-function allele.
Expression of fgf4, fgf24, and fgf17 in the distinct regions of pouches may suggest distinct roles for these genes in pouch formation. However, pouch formation was normal in single mutants for fgf4, fgf24, and fgf17 at 34 hpf, with the pouches being almost indistinguishable from those in wild types (Fig. 5A–D). Consistently, the development of hm and cb cartilages whose development relied on pouches was also normal in single mutants for fgf4, fgf24, and fgf17 at 5 dpf (Fig. 5I–L). To analyze a functional redundancy among the fgf genes in pouch formation, we examined all double mutant combinations for the fgf genes. However, pouches and facial cartilages, including hm and cb, were completely normal in all double mutant combinations (Fig. 5E–G, M–O), and they were also unaffected by the loss of all three fgf genes (Fig. 5H and P). We also examined facial bones in single, double, and triple mutants for the fgf genes, but they were indistinguishable from those in wild-type animals (red staining in Fig. 5I–P). Our result suggests that fgf4, fgf24, and fgf17 that are expressed in the pharyngeal endoderm in pouch formation, are highly redundant with other fgf genes in the development of pouches and facial skeletons.
DISCUSSION
In this study, we identified the expression of fgf4, fgf24, and fgf17 genes in the endoderm, with their regulation by Tbx1 and Fgf8a, in pouch formation. Their expression and regulation suggested that they might be necessary for pouch formation. However, functional analysis of the fgf genes did not support an essential role for these genes in pouch formation and facial skeletal development. It has been suggested that genetic redundancy in the Fgf signaling components generates a robust developmental system in zebrafish (Leerberg et al., 2019; Draper et al., 2003). Double mutants for fgf8a and fgf24 lead to loss of posterior mesodermal derivatives, whereas single mutants for fgf8a and fgf24 have normal mesoderm development (Draper et al., 2003). Similarly, while all single mutants for fgfr1a, fgfr1b, and fgfr2 genes are viable with no embryonic phenotypes, certain double and triple mutant combinations have defects in the brain, facial cartilages, pectoral fin, and posterior mesoderm (Leerberg et al., 2019). Considering the fairly normal expression of fgf4, fgf24, and fgf17 in fgf3 mutants, normal development of pouches and facial skeletons, even in the triple mutant for the fgf genes, could be due to their redundancy with fgf3 or other unidentified fgfs acting downstream of fgf3. To test this hypothesis, we need to analyze double and triple mutant combinations for fgf3 and fgf4, fgf24, and fgf17, as well as a quadruple mutant for the fgf genes. We predict that the defects in pouches and facial skeletons seen in those mutants would be similar to those seen in the animals blocking Fgf signaling in the pharyngeal endoderm. Alternatively, the triple mutant for fgf4, fgf24, and fgf17 shows normal development of pouches and facial skeletons due to a genetic compensation by other fgf genes, including fgf3 and unidentified fgf, expressed in pharyngeal endoderm. It has been shown that indel mutation generated by CRISPR/Cas9 system can lead to phenotypes that are weaker than either point mutation or morpholino-mediated knockdown due to a genetic compensation; in mutants, expression of a gene(s) related to the mutated gene is upregulated and functionally compensates for the mutated gene (Rossi et al., 2015; El-Brolosy et al., 2019). Although it is preliminary to identify novel fgf genes in pouch formation, our study still implies a potential role for fgf4, fgf24, and fgf17 in pouch formation. Currently, we are developing genetic tools to reveal the genetic mechanism for the high degree of redundancy of Fgf signaling in pouch formation.