
INTRODUCTION
Embryonic stem cells (ESCs) have been regarded as a useful tool to research embryogenesis at the cellular level and a promising tool for cell replacement therapy because of their unlimited proliferative properties and differentiation potential into all kind cell type of the body (Thomson et al., 1998; Doss et al., 2004; Nishikawa et al., 2007; Takahashi & Yamanaka, 2006).
Type I diabetes results from autoimmune destruction of β cells in the pancreatic islet. The destruction could be repaired by new β cell transplantation. It has been reported that cadaveric human islet transplantation to type I diabetic patients was effective to treat diabetes for 5 years (Bellin et al., 2012). However, this strategy has a limitation that islet donors are very scare. Thus, the derivation of β cells from ESCs that have an unlimitedly proliferating capacity could be an alternative to the preparation of a transplantable cell source for diabetic patients.
ESCs can be differentiated into pancreatic progenitors via the definitive endoderm with efficiencies (Kroon et al., 2008; Rezania et al., 2012). These cells can further be differentiated into functional β cells, insulin producing cells (IPCs) (Kroon et al., 2008; Rezania et al., 2012). ESCs can also be differentiated into IPCs via nestin-positive progenitor route (Lumelsky et al., 2001; Mao et al., 2009). The resulting IPCs from both protocols shared many similar features with pancreatic islet cells, but not mature, functional β cells (Wei et al., 2013).
SIRT1 is an NAD+-dependent deacetylase involved in numerous fundamental cellular processes including gene silencing, DNA repair, and metabolic regulation (Baur et al., 2010; Donmez & Guarente, 2010; Haigis & Sinclair, 2010). SIRT1 activity is inhibited by nicotinamide, which binds to a specific receptor site (Avalos et al., 2005). Nicotinamide has been known to maturate pancreatic progenitors from ESCs into IPCs. These suggest that control of SIRT1 activity affect the differentiation of ESCs into IPCs. Thus, in this study we examined whether SIRT1 knockdown affect the differentiation of human ESCs into IPCs.
MATERIALS AND METHODS
The human ESC line H9 (WiCell, WI, USA) were cultured on mitomycin C (10 μg/mL)-treated mouse embryonic fibroblasts in DMEM/F12 containing 0.1 mM β-mercaptoethanol, 1% NEAA, 0.1% penicillin/streptomycin, 20% knockout serum replacement (Invitrogen, Carlsbad, CA, USA), 1 mM glutamax (Gibco, Carlsbad, CA, USA) and 10 ng/mL basic fibroblast growth factor. H9 colonies were mechanically transferred every 4–5 days.
H9 was differentiated into IPCs via the definitive endoderm by the method described by Rui Wei et al. (2013) with some modification. hESCs of small clumps were plated on matrigel (1:50, BD Biosciences)-coated dishes and cultured with DMEM/F12 (Invitrogen, Carlsbad, CA, USA) supplemented with 100 ng/mL activin A (R&D), 1 μM wortmannin (Sigma, St. Louis, MO, USA), 1% N2 (Invitrogen) and 1% B27 (Invitrogen) for 4 days. The plated cells were induced into pancreatic progenitor cells under culture in IMDM/F12 supplemented with 2 μM retinoic acid (Sigma), 20 ng/mL fibroblast growth factor 7 (Peprotech, Rocky Hill, NJ, USA), 50 ng/mL Noggin (Peprotech), 0.25 μM KAAD-cyclopamine (Calbiochem, San Diego, CA, USA) and 1% B27 for 4 days. The pancreatic progenitor cells were expanded in high glucose DMEM (Welgene, Korea) supplemented with 50 ng/mL endothelial growth factor (Peprotech), 1% ITS (Sigma), and 1% N2 for 5 days. The pancreatic progenitor cells were developed into IPCs in low glucose DMEM (Invitrogen) /F12 (1:1) supplemented with 1% ITS, 10 ng/mL bFGF and 50 ng/mL exendin-4 (Sigma) for 9 days. The IPCs were maturated by detaching with 0.05% trypsin-EDTA and seeding to ultra-low attachment 6-well plates (Corning, Tewksbury, MA, USA) for 3 days.
IPCs were fixed in 4% paraformaldehyde in PBS for 20 min at room temperature. The cells were blocked for 1 hour at room temperature with 10% normal goat serum in PBS containing 0.1% Triton X-100. The cells were stained with primary antibodies and Alexa Fluor 488 or Alexa 594 nm-conjugated secondary antibodies in PBS containing 1% BSA and 0.3% Triton X-100. The nuclei was stained with DAPI. The following antibodies and dilutions were used : 1:100; mouse anti-pancreatic and duodenal homeobox 1 (PDX1) (Santa Cruz, Dallas, TX, USA), 1:100; rabbit anti-c-peptide (Cell signaling, Danvers, MA, YSA), 1:100, goat anti-Nkx6.1 (Santa Cruz), 1:100; TRITC-conjugated goat anti-rabbit IgG (Sigma), anti-goat IgG (Alexa fluorescence 488) (Abcam, Cambridge, UK), goat anti-mouse IgG (Alex fluorescence 594) (Abcam, Cambridge, UK) and goat anti-mouse (Alexa fluorescence 488) (Abcam, Cambridge, UK).
10 mg/mL Dithizone (DTZ, Sigma) in dimethyl sulfoxide (DMSO) was diluted to 10 μg/mL DTZ in PBS. IPC clusters were stained at 37℃ for 15 min with the solution.
Cells were lysed with M-PER mammalian protein extraction reagent (Pierce, Rockford, IL, USA) supplemented with a protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and a phosphatase inhibitor cocktail (Roche) on ice for 1 hour. After spin down (10,000×g, 10 min), the cell lysates were taken from the supernatant, separated by SDS-PAGE, and transferred to PVDF membrane at 60 V, 4℃, overnight. The membrane was blocked with 3% skim milk in tris-buffered saline containing 0.05% tween-20 for 1 h and incubated with anti-SIRT1 antibody for 3 h at room temperature. After washing, the membrane was further incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Proteins were analyzed using an enhanced chemiluminescence detection system (TaKaRa, Japan).
To establish stable H9 expressing control or SIRT1 specific shRNA, H9 human ESCs cells were infected with control or SIRT1 shRNA expressing lentiviral particles (Sigma) and selected in the presence of 1 μg/mL of puromycin for 1 weeks. The cells were maintained in the presence of 1 μg/mL of puromycin. The knockdown level of SIRT1 gene was analyzed by western blotting with anti-SIRT1 antibody.
Expression of V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), PDX1, insulin (INS), somatostain (SST) and glucagon (GCG) genes was analyzed by QRT-PCR. PCR primers for those genes were purchased from Genotech (Daejeon, Korea). Total RNA was prepared using RNA Extraction Kit (QIAGENE, Germantown, MD, USA). RNA samples were reverse transcribed with random primers using Reverse Transcription System (Power cDNA Synthesis Kit; QIAGENE). Real-time PCR was performed with C1000 thermal cycler (Bio Rad, Hercules, CA, USA) and SYBR Premix Ex Taq (Takara, Japan). Quantitation of expression levels of pancreatic genes was performed by a CFX96 Real-Time PCR Detection System (Bio Rad). Fold-difference was calculated using the ΔCt method normalized to GAPDH.
Data were expressed as the mean±SD of at least 3 independent experiments. The difference in means between 2 groups was analyzed using the Student’s t-test.
Primer sequences used in this study are listed in Table 1.
QRT-PCR, Quantitative real-time polymerase chain reaction; GCG, glucagon; SST, somatostain; INS, insulin; PDX1, pan-creas/duodenum homeobox protein 1; MAFA, v-maf musculoaponeurotic fibrosarcoma oncogene homolog A; GAPDH, glyceraldehydes-3-phosphate dehydrogenase.
RESULTS AND DISCUSSION
To examine whether SIRT1 affects the cluster size of human ESC-derived IPCs, we knockdowned SIRT1 by infecting human ESCs with control or SIRT1 shRNA expressing lentiviral particles. The SIRT1 expression level were decreased by the infection of SIRT1 shRNA expressing lentiviral particles (Fig. 1A). SIRT1 knockdown enhanced the cluster size of human ESC-derived IPCs by approximately 60% (Fig. 1B and 1C).
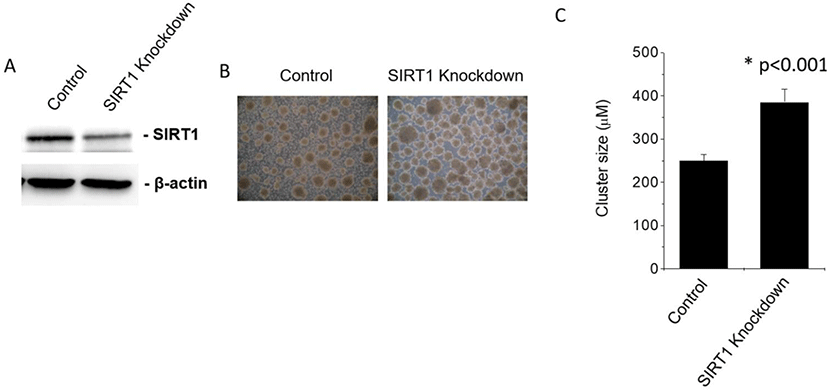
C-peptide is a substance produced by pancreatic β cells when proinsulin splits apart and forms one molecule of C-peptide and one molecule of insulin (Steiner et al., 1967). Since C-peptide and insulin are produced at the same rate, C-peptide is a useful marker of insulin production. We checked the effect of SIRT1 knockdown on c-peptide expression in human ESC-derived IPCs. Expectedly, SIRT1 knockdown enhanced the expression of C-peptide in IPCs differentiated from SIRT1-knockdowned human ESCs compared to those from control human ESCs (Fig. 2). These data suggest that the inhibition of SIRT1 activity enhances the differentiation of human ESCs into IPCs.
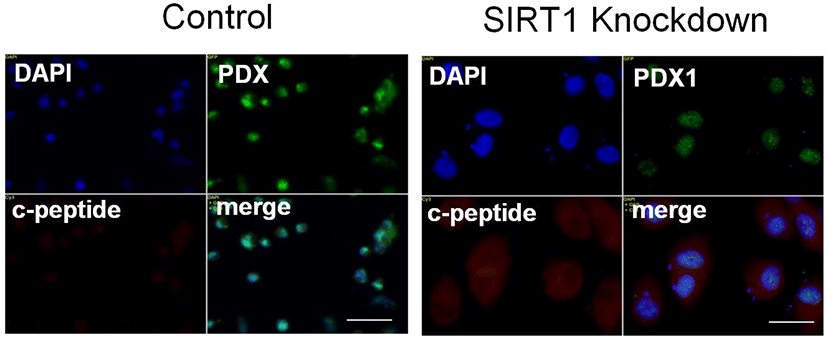
The homeobox transcription factor NK6 homeobox 1 (Nkx6.1) is essentially required for β-cell development (Sander et al., 2000). Nkx6.1 is highly expressed during a period in which β cells grow and differentiate (Sander et al., 2000), suggesting that Nkx6.1 expression may be involved in expansion of functional β-cell mass. Consistent with this idea, overexpression of Nkx6.1 in mature islets results in improved glucose-stimulated insulin secretion and enhanced β-cell proliferation (Schisler et al., 2008). We observed that the expression of Nkx6.1 in IPCs from SIRT1 shRNA expressing human ESCs is similar to that from control shRNA expressing human ESCs (Fig. 3). However, SIRT1 knockdown enhances and insulin in human ESC-derived IPCs (Fig. 3). These data are consistent with the data showing the enhancement of the cluster size and C-peptide expression of human ESC-derived IPCs.
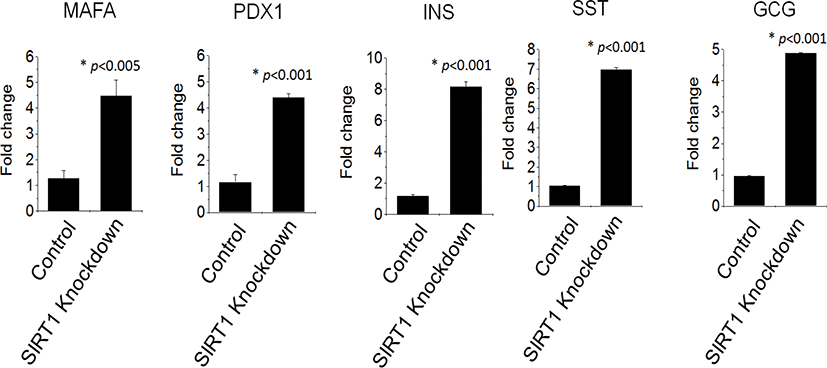
QRT‐PCR reaction, as shown in Fig. 4, demonstrated an enhanced expression of pancreatic genes in IPCs differentiated from SIRT1 shRNA expressing human ESCs compared to those from control shRNA expressing human ESCs. SIRT1 knockdown enhanced the transcription factors, PDX1 and MAFA, by four times in human ESC-derived IPCs (Fig. 4). The expression of the growth factors, glucagon, insulin and somatostatin, were highly enhanced in IPCs from SIRT1 shRNA expressing human ESCs compared to those from control shRNA expressing human ESCs (Fig. 4). These data suggest that SIRT1 knockdown enhances the functional differentiation of human ESCs into IPCs.
DTZ, a zinc-chelating agent, is known to selectively stain zinc in the insulin granule in pancreatic β cells (Latif et al., 1988). SIRT1 knockdown enhanced the staining intensity in the clustered IPCs derived from human ESCs (Fig. 5). These data suggest that SIRT1 knockdown enhances insulin contents in human ESC-derived IPCs.

SIRT1 catalyzes the deacetylation of acetyl-lysine residues of p53 and inhibits its ability to transactivate genes such as p21cip1/waf1 (Vaziri et al., 2001). SIRT1 inhibits apoptosis induced by exogenous H2O2 through inhibition of p53 function to activate expression of proapoptotic genes (Kume et al., 2006). However, it has been demonstrated that SIRT1 has different effects in response to ROS which depends on the severity of stress (Han et al., 2008). SIRT1 acts as an apoptosis inducer, instead of a survival factor, at physiological levels of ROS (Han et al., 2008). In this study, we found that SIRT1 knockdown enhances the cluster size of human ESC derived β cells. Thus SIRT1 knockdown-enhanced increase in the cluster size of human ESC-derived IPCs might be mediated by the anti-apoptotic effect of SIRT1.
It has been reported that SIRT1 positively regulates insulin secretion in pancreatic β cells (Bordone et al., 2006). SIRT1 represses the uncoupling protein (UCP) gene UCP2 by binding directly to the UCP2 promoter. SIRT1-mediated repression of UCP enhances ATP generation and increases insulin secretion by activation of voltage-gated K+ATP channels. Thus although SIRT1 gene deletion enhances β cell differentiation from human ESCs according to our data, there is a possibility that β cells derived by SIRT1 gene deletion has a defect in insulin secretion. Thus, we suggest that SIRT1 should be transiently inhibited during β cell differentiation from human ESCs.