
INTRODUCTION
Aquaporins (AQPs) are a family of small integral membrane proteins that primarily transport water across the cell membrane along osmotic gradients. Until now, 13 AQPs have been found in mammals (AQP0-12), some of which permit transcellular passage of glycerol and urea as well as water (AQP3, 7, 9, and 10) (Ishibashi et al., 2009; Verkman, 2011). AQP1, AQP3, AQP4, AQP8 and AQP9 have been reported to express in placenta and fetal membranes and suggested to play a key role in fetal fluid balance (Mann et al., 2002; Wang et al., 2006; Zhu et al., 2009). Mann et al. (2002) described increased amniotic fluid volume in AQP1 knock-out mice, speculating the association of AQP1 deficiency in fetal membranes with idiopathic polyhydramnios (Mann et al., 2005). Zhu et al. (2009) observed decreased expression of AQP1 and AQP3 in amnion of human term pregnancies with oligohydramnios as well as increased expression of AQP8 and AQP9 in polyhydramnios (Zhu et al., 2009; Jiang et al., 2012). Despite these reports, the precise role of AQPs in regulation of fetal fluid and electrolyte balance remains to be determined.
AQP3 is a water/glycerol transporting protein, which expresses in the basolateral membranes of epithelial cells in kidney collecting duct, airways, intestine and epidermis (Verkman, 2005). Mice lacking AQP3 manifest various degrees of nephogenic diabetes insipidus resulting from inability to concentrate the urine, and dry skin with decreased elasticity and impaired biosynthesis due to reduced glycerol and water content in epidermis (Ma et al., 2000; Hara et al., 2002). Based on the revealed function of AQP3 in other tissues, we speculated that AQP3 deficiency in placenta and fetal membranes might affect the fetal growth and development by changing the amniotic fluid volume and composition due to the defects in maternal-to-fetal transport of water and glycerol. However, the role of AQP3 in placenta has so far not been explored using mice lacking AQP3. Here, using AQP3 null mice, we showed that AQP3 deficiency in placenta and fetal membranes led to the intrauterine growth restriction.
MATERIALSA AND METHODS
AQP3 null mice generated by targeted gene disruption in embryonic stem cells in a CD1 genetic background were generously provided by Dr. Alan Verkman at University of California San Francisco, California, USA. The protocols for this study were approved by Dong-A University Medical School Institutional Animal Care Use Committee (DIACUC-07-20). Mice aged 8- to 12-week-old were used for this study. The presence of vaginal plug on the morning after mating was considered an evidence of successful copulation, and designated as 0.5 day post coitum (dpc). Timed pregnant mice were anesthetized and maternal blood was obtained from inferior vena cava. After the uterine horns were exposed by laparotomy, the whole gestational sacs were separated from the uterus, counted and weighed. Then, each gestational sac was isolated and weighed before and after aspiration of amniotic fluids. Amniotic fluid aspirated from 3 gestational sacs was pooled and stored at -70℃ for the chemical analysis. Finally, placenta, fetal membranes and fetuses were separated from each gestational sac and weighed individually. Amniotic fluid volume was calculated by subtracting the sum of weights of placental and fetal membrane, and fetus from that of a gestational sac.
Gestational sacs were fixed in 10% neutral buffered formalin overnight, paraffin-embedded and sectioned at 5 μm. For histological evaluation of placenta and fetus, the paraffin sections were routinely stained with hematoxylin and eosin. For immunohistochemistry, paraffin sections were deparaffinized, rehydrated and antigen-retrieved in citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 15 min at 100℃. After inactivation of endogenous peroxidase with 0.3% hydrogen peroxide, sections were incubated with 5% bovine serum albumin and 5% fetal bovine serum for 1 h at room temperature to block non-specific binding. Sections were incubated with primary antibodies; rabbit polyclonal anti-AQP1 (Chemicon, Temecula, CA, USA), anti-AQP3 (Chemicon, Temecula, CA, USA), and anti-AQP9 (Chemicon, Temecula, CA, USA) antibodies, overnight at 4℃. Subsequently, the sections were incubated with secondary anti-rabbit horseradish peroxidase linked IgG (Dako, Glostrup, Denmark) for 1 h at room temperature, and the reaction was visualized by DAB+ chromogen (Dako, Glostrup, Denmark). Tissue sections were mounted, and then visualized using digital scanning microscope (Scanscope, Aperio Technologies, Vista, CA, USA).
Frozen sections were air-dried, fixed in ice-cold acetone and blocked with 5% bovine serum albumin. The sections were incubated with anti-AQP antibodies as described above, and then with Alexa Fluor 488- or 555-conjugated anti-rabbit IgG antibodies (Molecular Probes, Eugene, OR, USA). The sections were mounted with Vectashield anti-fade reagent containing DAPI (Vector Laboratories, Burlingame, CA, USA) and visualized using confocal microscopy (LSM 510, Carl Zeiss, Thornwood, NY, USA) or fluorescence microscopy (IX70, Olympus, Tokyo, Japan).
Biochemical metabolic parameters in amniotic fluid and plasma were measured using commercially available kits according to the manufacturer’s protocols; for glucose by the hexokinase colorimetric assay kit (Sigma-Aldrich, Saint Louis, MO, USA), for glycerol by the glycerol assay kit (Sigma-Aldrich, Saint Louis, MO, USA), for triglyceride by the serum triglyceride determination kit (Sigma-Aldrich, Saint Louis, MO, USA), for lactate by the lactate assay kit (BioVision, Mountain View, CA, USA) and for FFA by the free fatty acid assay kit (Cell Biolabs, San Diego, CA, USA). Osmolality of serum and amniotic fluid was determined by freezing point method with Multi Osomometer (Precision System Inc., Natick, MA, USA). Electrolyte (Na, K and Cl) concentrations in amniotic fluid were determined by ion selective electrode method using Toshiba TBA 200FR (Toshiba Medical Systems Co., Ltd., Tokyo, Japan). Other chemical assays of amniotic fluids were done using automation system of diagnostic and laboratory medicine in Dong-A university.
RESULTS
In order to obtain the detailed information about the localization of AQP3 in placenta and fetal membrane, we first conducted immunohistochemistry and immunofluorescence using antibodies against different AQP subtypes. As shown in Fig. 1, AQP1 and AQP3 were abundantly expressed in placenta and fetal membrane showing a subtype-specific difference in subcellular localization. Anti-AQP3 antibody revealed strong labeling at the fetal-maternal interface in the labyrinthine region of placenta at E14.5 (Fig. 1A). AQP3 immunoreactivity was localized to the double-layered syncytiotrophoblasts of blood-placental barrier as well as the plasma membranes of fetal nucleated erythrocytes (Fig. 1B). The higher magnification image shows that AQP3 immunoreactivity was more restricted to the syncytiotrophoblast layer I (ST-1) facing maternal blood sinuses (Fig. 1D). In addition, AQP3 immunoreactivity was prominent in the basolateral membranes of visceral yolk sac endoderm as well as amniotic epithelium (Fig. 1C and E). However, no significant density of anti-AQP3 stain was detected in the mesodermal cells of visceral and parietal yolk sac. In contrast to AQP3, none of the three layers of trophoblasts composing the blood-placental barrier were immunoreactive for AQP1 (Fig. 1G and I). AQP1 immunoreactivity was detected at the plasma membranes of maternal rather than fetal red blood cells, and in endothelial cells lining both maternal and fetal blood vessels. In fetal membranes, AQP1 immunoreactivity was localized to the apical rather than basolateral membrane of visceral and parietal yolk sac endoderm as well as amnion (Fig. 1H and J).
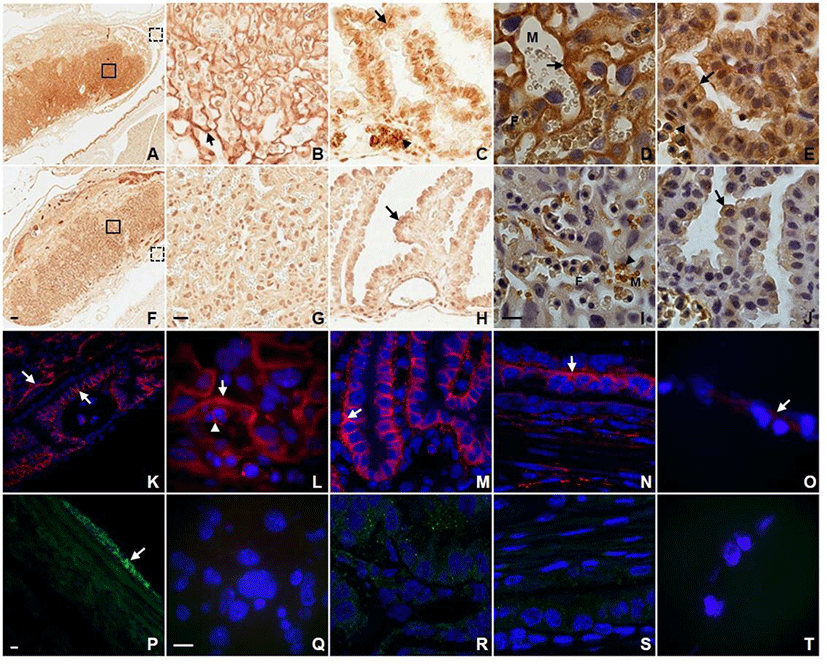
A distinct expression pattern of AQP3 in placenta and fetal membranes observed by immunohistochemistry was confirmed by immunofluorescence microscopy. An intense fluorescent anti-AQP3 signal was detected at the fetal-maternal interface of the labyrinth, the cell surface of fetal nucleated erythrocytes as well as the basolateral membrane of visceral yolk sac endodermal cells (Fig. 1K, L, M, and N). Amnion also displayed a low but detectable signal of anti-AQP3 (Fig. 1O). However, visceral yolk sac mesoderm, parietal yolk sac endoderm and endometrial epithelia were lack of AQP3 immunoreactivity. In contrast, AQP9 immunoreactivity was found neither at the fetal-maternal interface in the labyrinthine nor in visceral and parietal yolk sac (Fig. 1Q, R, S, and T). Only mesothelial cells of the perimetrium displayed a prominent AQP9 immunoreactivity (Fig. 1P).
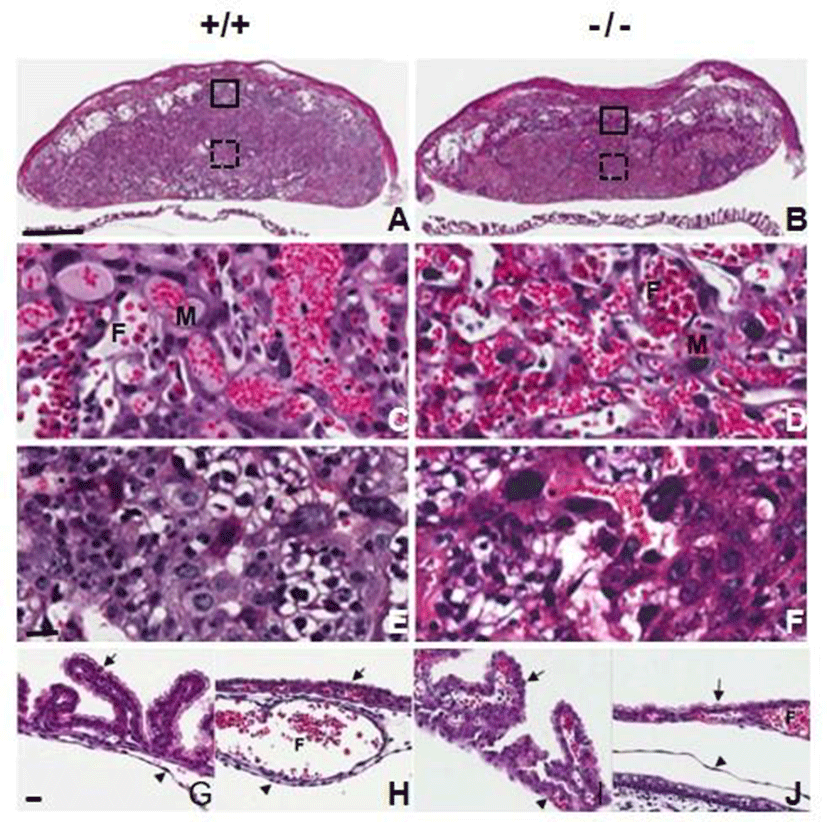
We examined whether AQP3 deficiency affects placental development through histological analysis of the placentas at E14.5 of AQP3 knock-out mice. Formation of chorioallantoic placenta appeared to be normal and three regions of decidua, the junctional zone and the labyrinth were well-recognized in AQP3-null mouse placentas as well as those of wild-type mice (Fig. 2A and B). Trophoblast giant cells, spongiotrophoblast cells and glycogen trophoblast cells were identified in the junctional zone, and three-cell-layered placental barrier developed normally in the labyrinth of AQP3-null mouse placenta (Fig. 2C, D, E, and F). In addition, the fetal membranes of AQP3 knock-out mice were also similar to those of wild-type mice; visceral yolk sac endoderm formed the villi near the placenta (Fig. 2G and H), and was transferred to microvilli-developed cuboidal cells of parietal yolk sac as it got farther away from placenta (Fig. 2I and J). There was no apparent histological defect observed during the development of placenta and fetal membrane due to AQP3 gene deficiency.
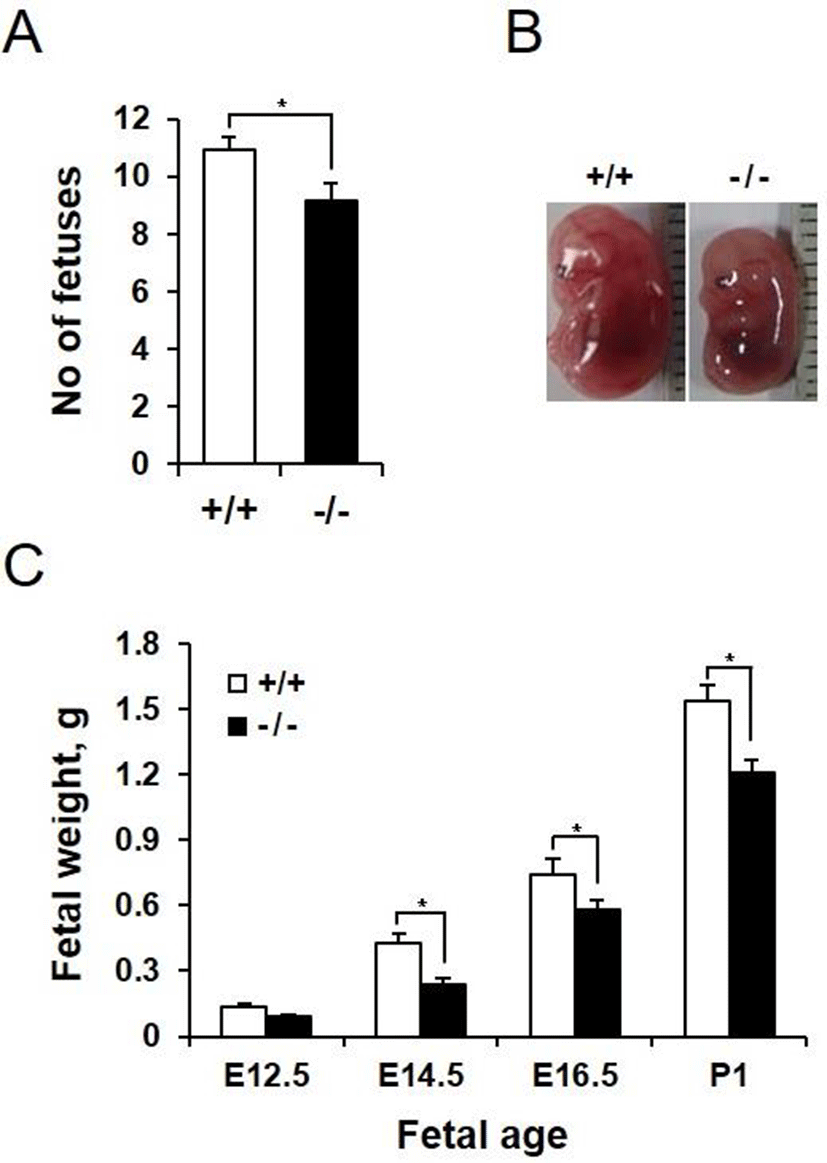
We next investigated whether AQP3 deficiency affects pregnancy and embryonic development. AQP3 knock-out mice had higher pregnancy failure rate than wild-type mice (67.21% vs 14.71%, data not shown). In addition, number of fetuses in a pregnancy was reduced in AQP3 knock-out mice compared with that of wild-type mice (9.13±0.54 vs 10.92±0.35, p<0.05, Fig. 3A). In spite of the small number of gestational sacs, fetuses of AQP3 knock-out mice were smaller than those of wild-type mice (Fig. 3B). The height of the fetus at E14.5, measured by the crown-rump length (CRL), was significantly reduced in AQP3 knock-out mice (10.5±1.1 mm vs 13.4±2.0 mm, p<0.05). AQP3 knock-out fetuses had lower body weight than wild-type fetuses regardless of the embryonic days during pregnancy. The difference in the weights between AQP3 knock-out and wild-type fetuses was highest on E14.5 (0.24±0.02 g vs. 0.43±0.03 g, p<0.05), showing a 44% reduction compared to the wild-type control (Fig. 3C). Although the difference in fetal weights reduced to 21% in the late gestation at E16.5, AQP3 knock-out fetuses still exhibited lower body weights at birth than did the wild-type fetuses (1.21±0.07 g vs. 1.53±0.09 g, p<0.05).
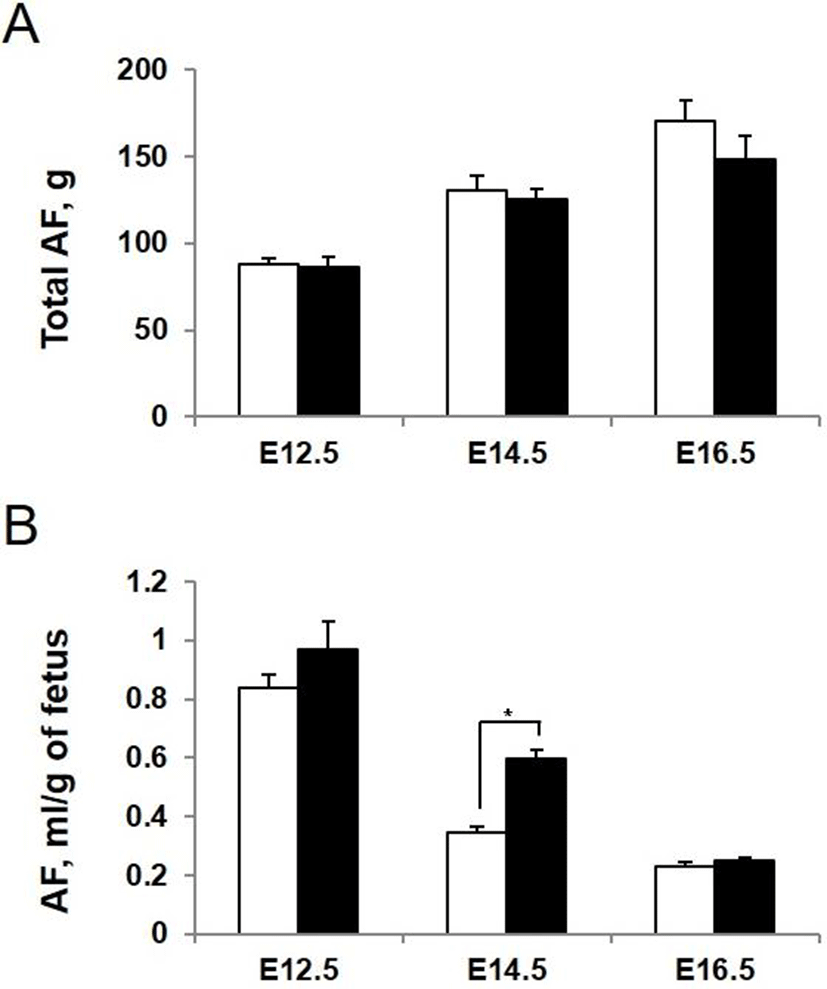
To determine whether AQP3 deficiency influences the amount and composition of amniotic fluid, we measured amniotic fluid volume directly at the different stages of development. The volume of amniotic fluid per gestational sac gradually increased during pregnancy. However, there was no significant difference in total amount of amniotic fluid in each gestational sac between AQP3 knock-out and wild-type mice (Fig. 4A). When the amniotic fluid volume was normalized to average weight of fetus considering the lower body weight of AQP3 knock-out mice than wild-type mice, it increased significantly during early to mid-gestation in AQP3 knock-out mice compared with wild-type mice (Fig. 4B). The biggest difference in amniotic fluid volume between AQP3 knock-out and wild-type mice was observed at E14.5 (0.60±0.02 mL vs. 0.35±0.03 mL, p<0.05), whereas the amniotic fluid volume of AQP3 knock-out mice became similar to that of wild-type mice during late gestation (E16.5).
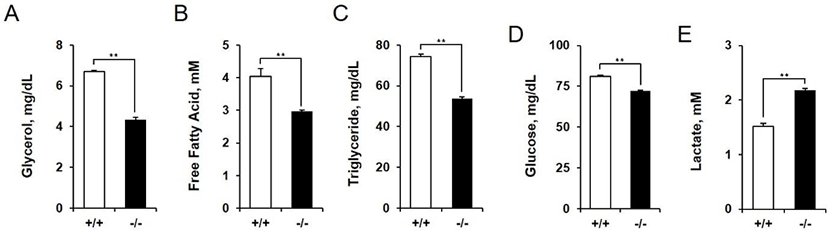
We measured the concentrations of metabolic substrates in amniotic fluid of AQP3 knock-out mice at E14.5. Concentrations of the metabolites measured were all reduced except lactate. Glycerol concentration in amniotic fluid showed the biggest difference between wild-type and AQP3 knock-out mice (6.69 mg/dL vs. 4.33 mg/dL, 1.54 fold decrease, p<0.01, Fig. 5A). The levels of triglyceride and free fatty acids in amniotic fluid were also low in AQP3 knock-out mice compared to wild-type mice (1.38 fold and 1.36 fold decrease, respectively, Fig. 5B and C). The glucose concentration in amniotic fluid was also decreased in AQP3 knock-out mice, but with a less difference between the two groups (81.10 mg/dL vs. 72.23 mg/dL, 1.12 fold decrease, p<0.01, Fig. 5D). However, the lactate concentration in amniotic fluid of AQP3 knock-out mice was much higher than that of wild-type mice (1.52 mM vs. 2.18 mM, 1.43 fold decrease, p<0.01, Fig. 5E).
DISCUSSION
We conducted this study in order to investigate the role of AQP3 in placenta and fetal membranes using AQP3 knockout mice. We found that AQP3 knockout mice exhibited intrauterine growth restriction as well as decreased metabolite concentrations in amniotic fluid. However, AQP3 knockout mice did not display hydroamnios or oligohydroamnios, although the amniotic fluid-to-fetal weight ratio increased in AQP3 null mice compared with wild-type mice.
The presence of AQP1, AQP3, AQP4, AQP8 and AQP9 transcripts and proteins have been described in placenta and fetal membranes (Liu et al., 2008; Hua et al., 2013). AQP1 expresses in the epithelial cells of chorionic plate amnion as well as endothelial cells in placenta and yolk sac of both human and mouse (Zhu et al., 2009). AQP3, AQP8 and AQP9 localize not only in epithelial cells of amnion but also in cytotrophoblasts and syncytiotrophoblasts of placenta in human (Damiano et al., 2001), whereas AQP4 is reported to express in stroma of human placenta (De Falco et al., 2007). In mice, AQP3 expression in amnion and trophoblasts of the labyrinth has been demonstrated (Kobayashi & Yasui, 2010). Here we first described the expression of AQP3 in the basolateral membranes of visceral yolk sac endoderm. We also determined the precise localization of AQP3 in the basolateral membrane of epithelial cells in amnion and the syncytiotrophoblast facing maternal blood sinuses in the labyrinth. In addition, AQP1 localization at the apical membrane of epithelial cells of both amnion and visceral yolk sac endoderm was also first described. Inconsistently with the previous reports (Damiano et al., 2001; Kobayashi & Yasui, 2010), AQP9 expression was not observed either in amnion or placenta except mesothelial cells of the perimetrium.
Development of placenta and fetal membrane is essential for normal fetal growth (Cross et al., 1994). Placenta provides vascular connections necessary for nutrient transport as well as attachment of the embryo to uterus. In mammals, two different types of placenta develop, yolk sac placenta (choriovitelline placenta) and chorioallantoic placenta (Carter, 2007). The yolk sac placenta formed in early pregnancy provides nourishment to the developing embryo until a definitive chorioallantoic placenta is established, then undergoing degeneration in human. Although striking differences in structure and function of the placenta are present among species, human and mouse placentas show structural similarities in terms of a highly invasive hemochorial type (Rossant & Cross, 2001; Cross et al., 2003; Enders & Carter, 2006). A comparative proteomic and transcriptomic investigation of placental tissues from both species revealed that over 80% of genes known to cause placental phenotypes in mouse are co-expressed in human (Cox et al., 2009). However, the number of trophoblast layers interposed between the maternal blood space and fetal capillaries is different between human and mouse. Human placenta is hemomonochorial with a single layer of syncytiotrophoblast adjacent to fetal endothelial cells, while the mouse placenta is hemotrichorial containing two syncytiotrophoblast layers and one cytotrophoblast layer in the placental barrier (Carter, 2007). We described here that AQP3 is strongly expressed in the syncytiotrophoblast in chorioallantoic placenta, an epithelium responsible for materno-fetal substrate transfer. Glucose (GLUT1 -3 and -4) and amino acid transporters (SNAT1, -2, and -4) have also been demonstrated to express at the microvillous and basal membranes of the syncytiotrophoblast in human placenta (Takata et al., 1994; Novak & Beveridge, 2000). Therefore, AQP3-mediated glycerol transport across the syncytiotrophoblast in placental barrier seems to be rate-limiting in embryo transfer and so do sugar and amino acids.
The yolk sac, which comprises both endoderm and mesoderm layers, is also a true placenta crucial for early embryonic development and survival in both human and mouse (Freyer & Renefree, 2008). The yolk sac serves as the only hematopoietic organ in early pregnancy and involves vasculogenesis, which is essential to fulfil its basic function as gas exchange and nutrient supply (Palis & Yoder, 2001). As the eutherian yolk sac essentially lacks nutrients inside the sac unlike that of an oviparous organism, maternal nutrients originating from the exocoelomic cavity or uterine glandular secretion are transferred to the growing embryo through the yolk sac. Uterine nutrients enter the yolk sac by passing through the trophoblasts, the parietal yolk sac endoderm cells and Reichert’s membrane formed between these cell layers vasculature (Sheng & Foley, 2012). Once inside the exocoelomic cavity, maternal nutrients are taken up by the visceral yolk sac endoderm and transported to the embryo through the vitelline. In the yolk sac-mediated route of nutrient transfer, the visceral yolk sac endoderm is the principal interface for nutrient uptake (Maurer & Cooper, 2005). Furthermore, the visceral yolk sac endoderm synthesizes cholesterol de novo and secretes serum proteins including apolipoprotein B (apoB)-containing lipoproteins, which play an essential role in lipid transport to the fetus (Plonne et al., 1992; Madsen et al., 2004). In this paper, we observed strong AQP3 expression in basolateral membrane of the visceral yolk sac endoderm, which is a key player in maternal-to-fetal nutrient transfer through yolk sac. Considering this with the finding of high AQP3 expression at the cell surface of fetal erythrocytes, we speculate that AQP3-mediated glycerol transport through yolk sac is highly activated in early pregnancy and presumably required for backbone of lipid or energy source to rapidly developing embryo.
Amniotic fluid provides an ideal environment for normal fetal growth (Brace, 1997). During mid to late gestation, amniotic fluid is produced from fetal urine and lung fluid, and removed by fetal swallowing and intramembranous absorption (Beall et al., 2007). AQPs in placenta and fetal membranes have been proposed to regulate the amniotic fluid volume. Alterations in their expression levels in placenta and fetal membranes have been reported to be related to polyhydramnios and oligohydramnios. It was demonstrated that AQP1 null mice exhibited polyhydroamnios and that AQP1 expression was increased in human placenta with idiopathic polyhydramnios (Mann et al., 2005). Alterations of AQP3, AQP8 and AQP9 expressions in fetal membrane were also reported in idiopathic polyhydramnios (Zhu et al., 2010). However, contrary to our assumption, we observed that AQP3 knock-out mice did not display polyhydramnios. We speculated that AQP3-mediated water transport across the basolateral membrane of amnion epithelial cells might be replaced with other AQPs present in the same membrane domain, whereas water reabsorption across the apical membrane of amnion epithelial cells and subsequently to blood vessels beneath through AQP1 cannot be substituted by any other AQPs. In addition, inability to concentrate the urine in AQP3 knock-out fetus did not seem to contribute to an increase in the amniotic fluid volume presumably due to no necessity for concentrating urine in fetal life floated in amniotic fluid.
Maternal triglycerides have been suggested as a primary source of fatty acids and glycerol because of their substantial increase in mid- to late gestation (Herrera, 2002). As maternal triglycerides cannot directly cross the placental barrier, they are either hydrolyzed into free fatty acids and glycerol by lipoprotein lipase (LPL) and endothelial lipase (EL) in microvillous membrane or taken up by syncytiotrophoblasts via a receptor-mediated endocytosis as incorporated into lipoproteins (Gil-Sanchez et al., 2011). FFA transport across the plasma membrane, apical or basal., occurs either by a simple diffusion through lipid bilayer or by facilitated diffusion using a fatty acid translocase (FAT/CD36), a fatty acid transport protein (FATP) and plasma membrane fatty acid-binding protein (FABPpm) for the uptake of long chain polyunsaturated fatty acids (Schaffer & Lodish, 1994). FAT/CD36, five members of FATPs (FATP1-4, and 6) and FABPpm have been identified in placental trophoblasts (Campbell et al., 1998).
Whereas considerable studies have been carried out on FFA transport across placenta, little attention was paid on the glycerol transfer from mother to fetus. Glycerol released along with FFA during triglyceride hydrolysis is either passively diffused across the cell membrane or transported by a facilitated diffusion through aquaglyceroporins to enter fetal circulation. Based on our findings of AQP3 localization in the syncytiotrophoblast of chorioallantoic placenta and basolateral membrane of endoderm of visceral yolk sac as well as low amniotic fluid glycerol concentration in AQP3 knock-out mice, we speculated that AQP3-mediated glycerol transport across the syncytiotrophoblast in placental barrier and endoderm of visceral yolk sac seems to be a rate-limiting step in glycerol transfer of embryo. Glycerol is used as a backbone of phospholipids or a source of energy during fetal development. Glycerol is a precursor of glucose and enters the gluconeogenic pathway as dihydroxyacetone phosphate in order to be converted into glucose (Klein et al., 1990).
AQP3 knockout mice exhibited intrauterine growth restriction along with decreased concentrations of metabolite substrates including glycerol in amniotic fluid. During embryonic development, especially the second half of gestation when energetic and biosynthetic demand increases rapidly to support the exponential rate of fetal growth, mobilization of maternal fat stores is accelerated (Herrera et al., 2006). Decrease in maternal glycerol delivery to fetus due to the AQP3 deficiency despite increasing demand during the second half of gestation might explain the reduced growth rate of AQP3 knockout pups to some extent.
In conclusion, this study is the first to demonstrate that AQP3 deficiency in placenta and fetal membranes is associated with reduced metabolite concentrations in amniotic fluid and impaired fetal growth. Our findings that AQP3 plays an important role in supplying glycerol from yolk sac and maternal blood to developing fetus in mice and that glycerol and its transporter, AQP3 might be implicated in normal fetal growth in human pregnancy.