
INTRODUCTION
Somatic cell nuclear transfer (SCNT) have been researched in different animal species, and many factors and technical methods for success have been established. However, SCNT still has limitations depending on the technical conditions such as embryo culture system, micromanipulation of embryo and donor cell, and activation (Choi et al., 2004; Niemann et al., 2008; Hwang et al., 2013). Activation of recipient cytoplasm is a crucial step in SCNT. Activation methods of embryo involve ethanol and calcium ionophore treatments, electric stimulus (ES) and so on. However, this activation method has physical and chemical effects on the SCNT embryos (Im et al., 2007) and can induce cellular stress such as reactive oxygen species (ROS) and endoplasmic reticulum stress (ER stress) (Hwang et al., 2012; Lee et al., 2018).
ER stress is an excessive accumulation of unfolded or misfolded proteins due to a variety of factors in the in vitro environment. Under ER stress conditions in SCNT embryos, embryonic metabolism degeneration and apoptosis by embryo fragmentation can occur (Zhang et al., 2011; Lee et al., 2018). In the previous experiment, the generation of ER stress according to the activation method of embryo was confirmed, and it was occurred that the high levels of ER stress and cell damage in the treatment of Ca-ionophore (A23187) after ES (Park et al., 2017).
ER stress inhibitors such as tauroursodeoxychloic acid (TUDCA; Xie et al., 2002; Ozcan et al., 2006) and salubrinal (Boyce et al., 2005) are generally used to suppress ER stress. TUDCA is a known ER stress inhibitor as a chemical chaperone that stabilizes the structure of proteins (Xie et al., 2002). Salubrinal operate directly on the UPR signal, a defense system by ER stress, to increase ER chaperon (Boyce et al., 2005).
In this study, we investigated the effect of ER stress inhibitor treatment during parthenogenetic activation of oocytes on the ER stress generation, apoptosis, and in vitro development of parthenogenetic porcine embryos.
MATERIALS AND METHODS
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise indicated.
Collecting oocyte complexes (COCs) from porcine ovaries obtained from local slaughterhouses by aspiration from the follicles (3- to 6-mm diameter) using a 10 mL syringe with 18-gauge needle. After selecting quality COCs, they were incubated in Tissue Culture Medium 199 (TCM199; Gibco, Grand Island, NY, USA) supplemented with 0.1% polyvinyl alcohol (PVA), 3.0 mM D-glucose, 0.9 mM NA-pyruvate, 75 μg/mL penicillin G, 50 μg/mL streptomycin, 0.57 mM cysteine, 10 μg/mL epidermal growth factor (EGF), 0.01 IU/mL luteinizing hormone (LH), and 0.01 IU/mL follicle-stimulating hormone (FSH) at 39℃, 5% CO2 in air for 42–44 h.
The cumulus cells of matured oocytes were removed by eliminated in PBS supplemented with 0.1% (w/v) hyaluronidase and 0.1% (w/v) PVA for 2 min 30 sec. The oocytes were activated by two activation methods; 1) ES with the same condition as our fusion condition for SCNT experiment (E), and 2) ES followed by additional treatment with 10 μM Ca-ionophore (A23187) for 5 min (EC). For ES, two direct-current (DC) pulses of 1.25 kV/cm were applied for 30 μsec using a BTX Electro Cell Manipulator 200 (BTX, San Diego, CA, USA). After activation, parthenogenetic embryos were cultured in PZM-3 medium with or without (control) 200 nM salubrinal (Sal), 100 μM TUDCA or both salubrinal and TUDCA (Sal+TUD) at 39℃, 5% CO2 in air for 3 h. The concentration of ER stress inhibitor used in this study was previously determined using porcine somatic cells (Park et al., 2018).
After ER stress inhibitor treatment, parthenogenetic embryos were cultured in fresh PZM-3 medium at 39℃, 5% CO2 in air for 6 days. At the 1-cell stage (20 h) or blastocyst stage (Day 6), the embryos were lysed by using 20 μL Lysis/Binding (L/B) buffer of Dynabeads(r) mRNA Direct kitTM (Life Technologies, Oslo, Norway). The lysed samples were stored at -70℃ until use.
The mRNAs of parthenogenetic embryos at 20 h (1-cell stage) and Day 6 (blastocyst) after ES were isolated using the Dynabeads(r) mRNA Direct kitTM (Life Technologies) according to the manufacturer’s protocol. After thawing the embryo samples, 30 μL of Dynabeads oligo (dT)25 was added to each sample and shake for 8 min at room temperature to induce the hybridization of poly(A) tail of mRNA with oligo (dT)25 on the beads. The bead-mRNA complex was washed twice with buffer A and B, then the poly (A) mRNA was eluted from beads by incubation with 12 μL elution buffer (10 mM Tris-HCl) for 5 min at 75℃. The 6 μL of isolated mRNA was transferred to 0.2 mL micro-tube. The mRNA was reverse transcribed into cDNA for next analysis at 4℃ for 5 min, 37℃ for 20 min, 50℃ for 5 min, 98℃ for 5 min. For genomic DNA elimination and reverse transcription, 2 μL of 4x DN Master Mix and 5x DN Master Mix II were added to reaction tube at the specified time. The synthetic DNA products were stored at 4℃ until use.
The expression of x-box binding protein 1 (Xbp1) mRNA was detected by RT-PCR analysis. The expressions of four ER stress-associated genes, C/EBP homologous protein (CHOP), binding protein (BiP), activating transcription factor 4 (ATF4), and glucose-regulated protein 94 (GRP94), and two apoptotic genes, Bcl2-associated X protein (Bax) and caspase-3, were analyzed by RT-qPCR. For RT-PCR, the cDNA samples were reverse-transcribed according to the manufacturer’s instructions using AccuPower(r) Taq PCR PreMix (Bioneer, Daejeon, Korea). Initial denaturation was performed for 1 min at 72℃, followed by 36 cycles of amplification. Each cycle was composed of denaturation at 95℃ for 30 sec, annealing at 58℃ for 30 sec, and extension at 72℃ for 50 sec. Final extension was performed at 72℃ for 5 min to complete reaction. PCR products were analyzed by UV irradiation with the Gel DocTMXR+ (Bio-rad, Berkeley, CA, USA) on 1.2% or 4% agarose gel (Amresco, Cleveland, OH, USA) stained with ethidium bromide (Bioneer). RT-qPCR was performed using 1 μL cDNA with the power SYBR Green PCR master Mix (TOPrealTM qPCR 2X PreMIX; SYBR Green with high ROX, Enzynomics, Daejeon, Korea) in a StepOne Plus instrument (Applied Biosystems, Foster City, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control for the normalization of target gene expression. The Primer sequences of each gene were shown in Table 1.
Each activation method was replicated at least 3 times. Data of RT-PCR and RT-qPCR were analyzed by the analysis of variance (ANOVA) and Duncan’s multiple range test. Developmental rate was analyzed by Chi-square test, and the blastocyst cell number was analyzed by Student t-test. All analyses were performed using the Statistical Analysis System software package (v.9.4, SAS Institute, Cary, NC, USA).
RESULTS
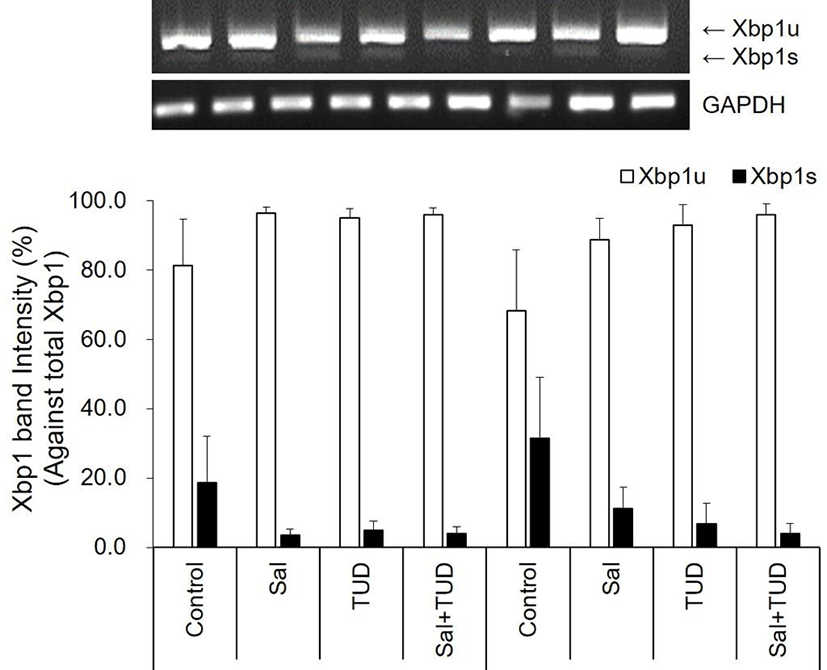
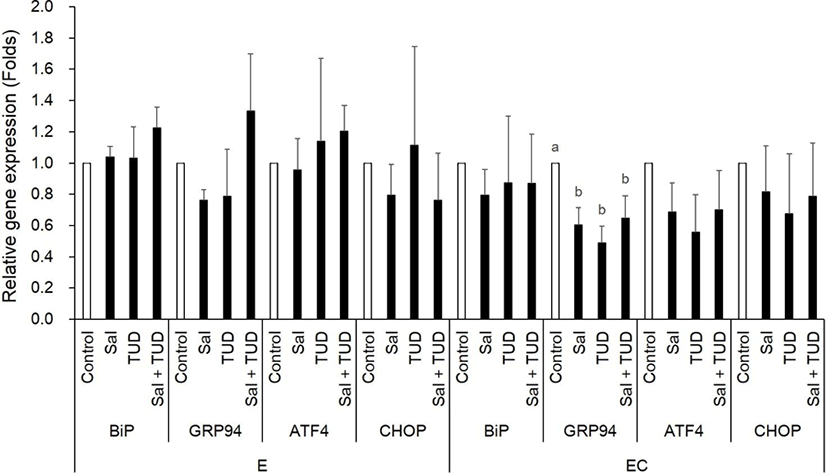
At the 1-cell stage, although Xbp1 splicing was slightly reduced by ER stress inhibitors, but no difference was observed among treatments in both E and EC group (Fig. 1). However, BiP transcription level in the E group was significantly reduced by salubrinal treatment, and GRP94 and ATF4 transcription levels in EC group were significantly reduced by all treatments (p<0.05) compared to control (Fig. 2).
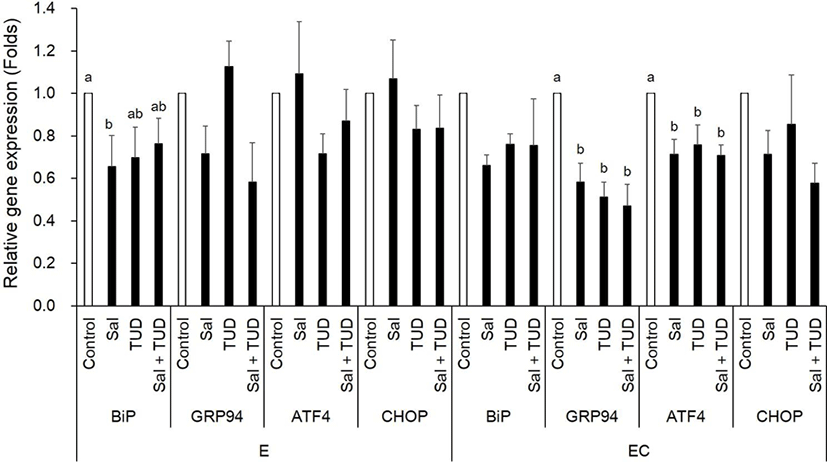
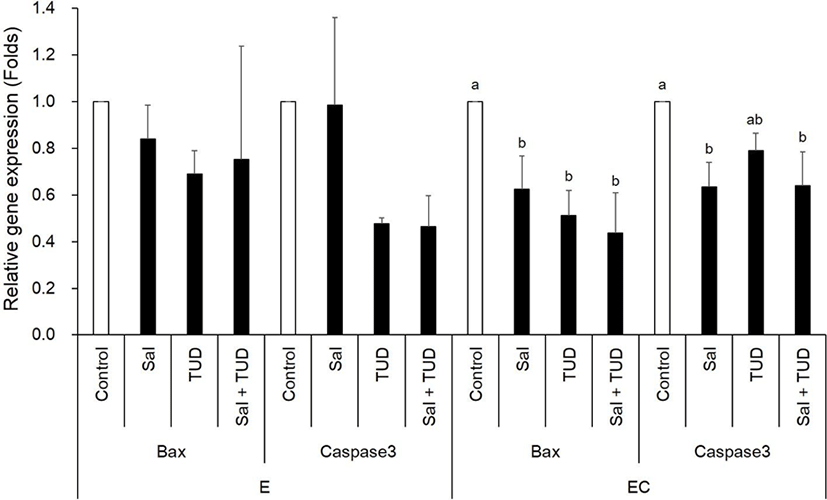
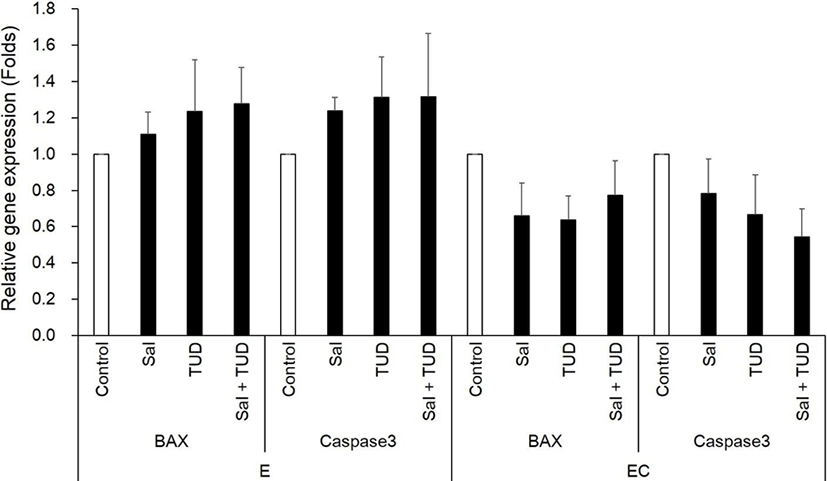
In the E group, there were no significant differences in the transcription levels of apoptotic genes (Bax and caspase-3) among treatments (Fig. 3). In the EC group, however, both apoptotic genes were reduced by ER stress inhibitor treatments compared to control (p<0.05) except Caspase-3 gene by TUDCA treatment.
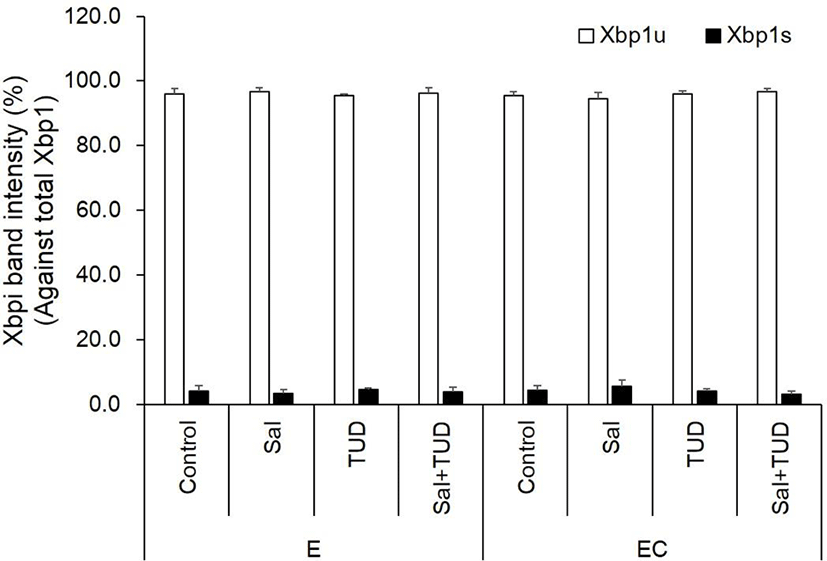
There were no significant differences in Xbp1 splicing and ER stress-associated genes among all treatments and control groups in the E and EC groups (Fig. 4 and 5) except Grp94 transcription level in the EC group, which significantly reduced compared to control by ER stress inhibitor treatments (p<0.05). There were no significant differences in the transcription levels of apoptotic genes (Bax and caspase-3) among treatments in both E and EC groups (Fig. 6).
Blastocyst formation rate was significantly improved in the E group by TUDCA or salubrinal treatment (22.3% and 22.8%, p<0.05) and in the EC group by TUDCA treatment (21.5%, p<0.05). Blastocyst development was not improved by Sal+TUD in both groups (Table 2). The mean cell number in blastocysts, however, was significant increased in all treatment groups (47.4±6.2 to 66.1±5.3 in E group, and 63.9±8.2 to 74.4±2.8 in EC group) compared to the control (p<0.05).
Experiments were repeated 8 times in each treatment group.
E, electric stimulus; EC, E+10 μM Ca-ionophore (A23187) treatment; Sal, 200 nM salubrinal; TUD, 100 μM TUDCA.
DISCUSSION
A23187 activates various metabolism of oocytes by increasing intracellular calcium concentration and is used as one of the activation agent. However, when intracellular calcium concentration increases, ER stress homeostasis is destroyed and caused ER stress. When the UPR response is triggered by ER stress, the excess Ca2+ is released by activating CHOP protein, IRE1 mediator ASK1 / JNK in the ER, thereby increases the mitochondrial Ca2+ concentration (Yoshida, 2007; Sano & Reed, 2013). It activates apoptosis by increasing the expression of pro-apoptotic protein Bax and caspase-3 (Song, 2012; Sano & Reed, 2013). In the previous study, ER stress and apoptosis according to the activation method of porcine parthenogenetic embryos were analyzed, and it was confirmed that ER stress generation and apoptosis were increased in the treated with A23187 (Park et al., 2017).
In this experiment, the salubrinal and TUDCA were used as an ER stress inhibitor to inhibit ER stress and apoptosis caused by activation treatment. It was reported that the TUDCA decreases ER stress-induced apoptosis by regulating intracellular Ca2+, and maintain homeostasis using intracellular free Ca2+ (Beuers et al., 1993a,b; Xie et al., 2002). In addition, TUDCA and salubrinal have been reported to prevent ER calcium depletion which cause ER stress (Xie et al., 2002; Boyce et al., 2005).
In this study ER stress inhibitor treatment did not affect the ER stress and apoptosis when oocytes were activated by ES. Contrastively ER stress inhibitor decreased the expression of GRP94, ATF4 and apoptotic genes in the EC group at the 1-cell stage. It is suggested that ER stress inhibitor can reduce the ER stress and apoptosis of parthenogenetic embryos induced by A23187 (Park et al., 2017).
In this study, the effect of ER stress inhibitor was marginally in XBP1 splicing, ER stress-associated genes and apoptotic genes in the blastocyst stage. In the previous study, when TUDCA was treated for culture period in the SCNT embryos, ER stress and apoptosis were reduced in the blastocyst stage (Zhang et al., 2011, 2012). ER stress inhibitor treatment during the culture period may have continued to influence the expression of genes in blastocyst. In this study, however, ER stress inhibitor treatment was performed only for activation period, not culture period. A previous our study also showed that the expression of genes in blastocyst stage was minimal when ER stress inhibitors were treated only during the micromanipulation and activation period in the SCNT embryos (Park et al., 2018). In this experiment, the ER stress induced in the early development stage was suppressed by short-term treatment only during the activation period.
In this study, blastocyst formation rate tended to increase when treated with salubrinal or TUDCA alone, but not by the combination treatment of salubrinal and TUDCA. Similar trend was observed our SCNT study (Park et al., 2018). It seems that combined treatment of both ER stress inhibitors may have an excessive impact on the embryos. However, when the ER stress inhibitor was treated simultaneously as well as separately, the number of cells was significantly increased in all treatment. This mean that the ER stress inhibitor (salubrinal and/or TUDCA) treatment may also have an effect on quality of parthenogenetic embryos.
In conclusion, these results suggest that the treatment of ER stress inhibitor during parthenogenetic activation can reduce ER stress, and thereby reduce apoptosis and promote in vitro development of porcine parthenogenetic embryos.