
INTRODUCTION
Histiotypic neoplasia in Osteosarcoma (OS) is one of the most common malignant primary bone tumor specifically in the bones of fore limb, hind limb and metaphysical region of long bones (Longhi et al., 2006; Vigorita, 2008). Damage to the interconnecting soft tissues is a major concern with such kind of tumors (Vigorita, 2008). It is one of the highly metastasized cellular tumors with pleomorphic spindleshaped cells. These spindle shaped cells are involved in the efficient generation of an osteoid, osteoclast and osteoblast matrix and very well histologically characterize the OS (Vigorita, 2008). It is reported to have high incidence in adolescents and young ones. 60–70% of survival rate up to five years after diagnosis has been reported in the patients with 8.9% mortality in children (Longhi et al., 2006). OS is reported to have moderate morbidity (10 to 26 cases/ million/year) (Stiller, 2007). The metastasized cells most often spread to the lungs, bones and brain (Longhi et al., 2006). The multi-agent approach involves chemotherapy, surgery and radiation but it is not fully adequate to eliminate OS (ACS, 2013). Longhi et al. (2006) and NIH (2014) reported, prototype OS manifests few frequent traits like growth rate, cell division with aggression and early pulmonary metastases. Furthermore, Karlsson et al. (2013) reported 33 high inheritable loci for OS in multiple dog breed by genome-wide association study (GWAS). Collectively, these data show the dire need of an essential innovative drugs and approaches to further improve the prognosis of OS patients.
Bio-signal trafficking is the pivotal functional mechanism acquired in the differentiation, proliferation, growth and survival of the cells (Lin & Liu, 2008). During histeotypic neoplasia normal cells follow altered ways to proliferate independently due to growth promoting conditions (Iliopoulos et al., 2009). At the same time, growth inhibitory signals with respect to metastasis, elicit an angiogenic responses as well as to evade mechanism that arrest cell growth cycle and proliferation (Martin, 2003). Inhibition of kappa B kinase blocks the induction of nuclear factor-kappaB (NF- κB) signaling and makes bridge between inflammation and cancer (Lin & Liu, 2008; Oeckinghaus et al., 2011). Ghosh & Karin, (2002) reported regulation of the wide variety of genes encoding cytokines (e.g., IL-1, IL-2, IL-6, IL-12, TNF-α, LTα, LTβ and GM-CSF), chemokines (e.g., IL-8, MIP-1α, MCP1, RANTES and eotaxin), adhesion molecules (e.g., ICAM, VCAM and E-selectin) acute phase proteins (e.g., SAA) and inducible effector enzymes (e.g., iNOS and COX-2) by NF-κB. Moreover, consequently NF-κBdependent transcription is not only tightly controlled by positive and negative regulatory mechanisms but also closely coordinated with other signaling pathways such as JNK, Akt and MAPK (Baker et al., 2011; Oeckinghaus et al., 2011).
Small non-coding microRNA (miRNA) is a primary regulator of signal trafficking processes which modulate bio-physiological activities such as developmental timing, proliferation, cell metabolism, differentiation and cell apoptotic activity (Mendell & Olson, 2012). The modulation takes place by the suppression of several candidate genes during transcription and post-translation on the basis of ambiance. The abnormal expression of miRNA observed in number of cancer patients presents miRNA as candidates targets for the therapeutic intervention of neoplasm including OS (Mitchell et al., 2008; Davis & Hata, 2009). The transcriptional regulation of miRNAs occurs near the intron region of protein coding and non-coding genes by their host gene promoter. It is also reported that RNA-induced silencing Complex (RISC) negatively suppresses the gene expression of targeted mRNA through the 3´ untranslated region (3´ UTR) (Davis & Hata, 2009). The miRNA plays a dual role in cancer biology. It can either induce or suppress the proliferation or both (Esquela-Kerscher & Slack, 2006). The miRNA works as potential biomarkers and therapeutic targets in different sarcoma including OS (Ouyang et al., 2013). Furthermore, the miRNA based approaches for eradication and diagnosis of OS can be handled in two strategic ways (Kobayashi et al., 2012) i.e. to inhibit the expression of oncogenic miRNA using antisense oligonucleotides (anti-miR) and secondly to induce the expression level of tumor suppressor miRNAs by inserting miRNA mimics (Esquela-Kerscher & Slack, 2006; Shimada, 2013). A noble approach of study has revealed by Poos et al. (2013) that miRNAs and transcription factors (TFs) are master regulators of the biological systems. In contrast, miRNAs and TFs together make mega diverse network to have desired action and finally results in distinct cellular phenotype. It is reported here that several genes, TFs, and miRNAs form co-regulatory network in OS cell growth as one hallmark in cancer (Poos et al., 2013). Therefore, at this point the understanding of the role and mode of action of miRNAs is need of hour.
In this study, an attempt has been made to correlate several miRNAs with different factors of nuclear factor of kappa B (NF-κB). A novel bridge model between “miRNAs and NF-κB” has been proposed in the current study to have an efficient control of the OS. The miRNA profiling of primary tumors has prognostic value for the survival rate and it targets the beneficiary results for OS patients who previously had third to sixth-line treatment with anti-epidermal growth factor receptor (EGFR) based therapy.
NF-κB VERSUS THE IκB FAMILY AS MASTER REGULATOR
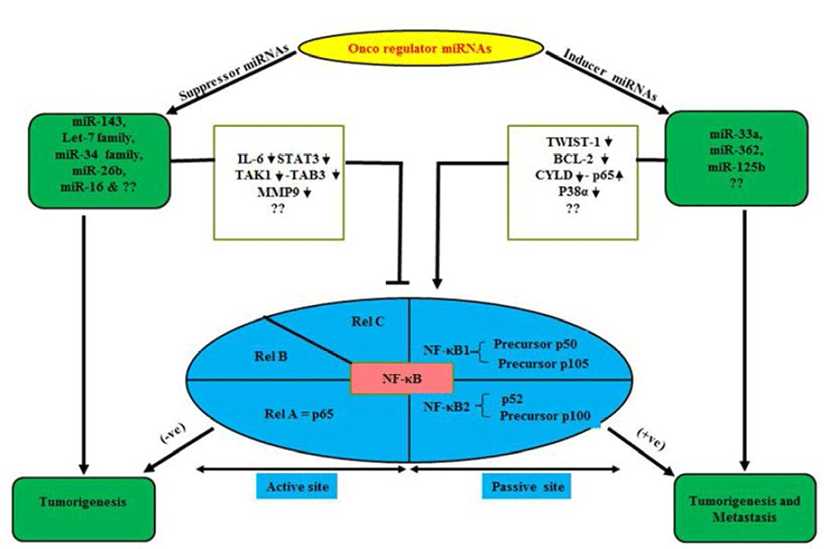
NF-κB plays a central role in the regulation of diverse biological processes, including immune responses, development, cell proliferation and survival (Esquela-Kerscher & Slack, 2006; Perkins, 2007). At the same time deregulated NF-κB has been linked to a variety of human diseases; in particular cancers (Perkins, 2007; Karin, 2009). The NF- κB, a transcriptional regulator consists of reticuloendotheliosis (Rel) protein dimers and binds with a DNA sequence motif known as κB site (Huxford & Ghosh, 2009). Depending upon their requirement for proteolytic processing the Rel protein family is classified into two groups (Huxford & Ghosh, 2009). The Rel proteins which require proteolytic processing includes NF-κB1 (also known as p105) and NF-κB2 (p100), which are processed to produce the mature p50 and p52 proteins, respectively (Fig. 1). The Rel protein without proteolytic processing includes Rel A (also known as p65), Rel-B and Rel-C. NF-κB signal cascade involves canonical and non-canonical pathways (Sun, 2011). It is worth mentioning over here that canonical pathway induced by extracellular factors includes tumor necrosis factor α (TNF-α) and Interlukin-1 (IL-1) those activate NF-kappa-B essential modulator (NEMO) (Oeckinghaus et al., 2011). Further it has been reported that NEMO makes complex with Inhibitor kappa kinase (IKK) α/β and p50/ RelA to phosphorylate the Inhibitor of kappa B alpha (IkBα). Later, phosphorylated p50/RelA translocates to nucleous to activate target genes (Ghosh & Karin, 2002). Similarly, Lipopolysacharide (LPS) and CD40L are reported to activate NF-inducing kinase (NIK) to initiate noncanonical pathway by involving IKKα/β, p100, RelB complex and phosphorylated p100 activate RelB/p52 (Sun, 2011) which translocate to nucleous and interact with target κB element and regulate transcription (Lin et al., 1998). Receptor activator of nuclear factor kappa-B kinase (RANK) in osteoclast formation and bone remodeling via TRAF, regulate seven different signaling pathways including four directly (IKK-NF-κB, JNK1, Myc, calcineurin-NFAT mediate osteoclastogenesis), two (Src-Akt, p38) by osteoclast activity and two other promote osteoclast survival via Src- Akt and Erk1/2 (Beristain et al., 2012).
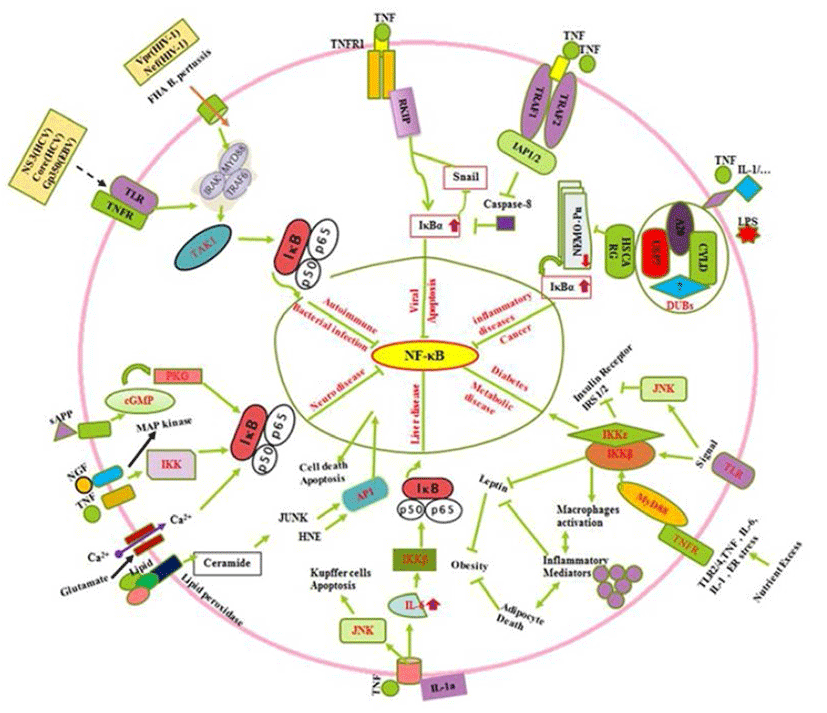
All known NF-κB activators converge toward the IKK complex to activate the canonical and non-canonical NF- κB pathways (Sun, 2011). The IKK complex contains two catalytic (IKKα and IKKβ) and a regulatory subunit (NEMO/IKKγ) which those regulate the canonical NF-κB pathway, whereas IKKα regulates the non-canonical pathway (Oeckinghaus et al., 2011). The process of IKKα activation and its role in the regulation of canonical NF-κB activation remain elusive (Hoesel & Schmid, 2013). In the canonical NF-κB signaling pathway, stimuli such as tumor necrosis factor α (TNFα) recognition by the TNF receptor leads to activation of the inhibitor of IkBa kinase (IKK). IKK then phosphorylates IκBα, which results in K48-linked polyubiquitination of IκBα followed by proteasome-dependent degradation (Oeckinghaus et al., 2011). It can regulate the Raf-1 kinase inhibitor protein (RKIP) by the down regulation of zink finger protein (SNAIL) during the cytotoxic T-cell response to viral infection and/or cancer in cells expressing the resistant NF-kappaB-Snail-RKIP phenotype (Fig. 2) (Baritaki & Bonavida, 2010). Moreover, Ubiquitin is a small protein that exists in all eukaryotic cells where it can attach to lysine residues of substrate protein in posttranslational modification with the help of E1s, E2s and E3s enzymes to form Lysine 48-linked chains to proteasome that degrade and recycle the unwanted proteins (Rieser et al., 2013). Furthermore, NF-κB can be downregulated by cysteine methylation mediated disruption of Ubiquitin chain via E. coli NIeE, S-adenosyl-L-methionine-dependent methyltransferase activity in the the Npl4 zinc finger (NZF) domains in TAB2 and TAB3 (Zhang et al., 2012). The IKK family and their regulatory subunit NF-kappa-B are essential modulators (NEMO, also known as IKK-γ) which have ubiquitin-binding domain that binds to polyubiquitin chains to form a huge complex. It facilitates the interaction between IKKβ and its kinase TGF-β-activated kinase (Wu et al., 2006). In case of Crohn’s disease, nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is reported to induce NF-κB activation via K63-linked ubiquitin conjugation to NEMO (Abbott et al., 2004). The human protein HSCARG (also termed as NmrA-like family domain containing 1), several Deubiquitinases (DUBs), protein A20, cylindromatosis gene (CYLD), Ubiquitin specific proteases 7 (USP7) and several unknown enzymes interact to form complex which can inhibit the NEMO polyubiquitination. Inhibition of NEMO polyubiquitination is reported to decrease proteasomal degradation of IκBα (Li et al., 2014), which retains NF-κB in the cytoplasm. This complex further suppresses NF-κB activity (Fig. 2) (Pujari et al., 2013). Oeckinghaus et al. (2011) reported the activation of NF-κB via the binding of a ligand e.g., TNFα, IL-1, TNF superfamily of molecule ligand (CD40L). Conjugation of LPS to its receptors is also reported to activate binding of TNF receptor type 1-associated DEATH domain (TRADD) to TNF receptor-associated factor 2/Receptor-interacting serine/threonine-protein kinase 1 (TRAF2/RIP1) (Solt et al., 2007). It further triggers a series of events involving signaling cascade (Fig. 2) through phosphorilisation of IKKα/β (Lin et al., 1998). The activation of NF-κB by TNFα is strongly linked with the inhibition of apoptosis due to the ability of NF-κB to regulate expression of antiapoptotic genes such as TRAF1, TRAF2, c-IAP1, cIAP2, Bcl-2, and Bfl-1/A1 via suppression of Caspase-8 (Fig. 2) (Wang et al., 1998). In addition, extracellular factors including TNF, NGF, sAPP, lipids and minerals such as Ca2+ up regulates the IκBα by IKK activation (Mattson & Camandola, 2001), by induced cyclic GMP that activate cGMP-dependent protein kinase (PKG) in neuronal abnormalities (Fig. 2). Collectively, it is observed that diverse signals (TNF-α, sAPPα, NGF, glutamate, increase in levels of intracellular Ca2+ and reactive oxygen species such as H2O2) can suppress NF-κB activation via phosphorilization of IκBα (Mattson & Camandola, 2001; Kaltschmidt et al., 2005; Kaltschmidt & Kaltschmidt. 2009). Several signal (prostate apoptosis response-4 {Par-4} and the lipid peroxidation product 4-hydroxynonenal {HNE}) those inhibit NF-κB activity is generated in neurons undergoing apoptosis (Mattson & Camandola, 2001). NF- κB is modulated by signals (Fig. 2) emanated from activator protein-1(AP1), glucose-regulated protein-78 (GRP-78), Jun NH2-terminal kinase (JUNK), cyclic guanosine monophosphate (cGMP)-dependent protein kinases (PKGs) and cGMPdependent protein kinase (Mattson & Camandola, 2001). Moreover, TLR2/4, TNF, IL-6, IL-1 and endoplasmic reticulum (ER) stress during metabolic imbalance show insulin resistance which later activate cytokine inflammatory as well as insulin regulating trafficking by Toll-like and cytokine receptors through MyD88 (Baker et al., 2011). In addition the death domain adaptor molecules via TRAFs result in phosphorylation and activation of the IKK complex which control the diabetogenesis (Fig. 2) (Chen, 2005). It leads to activated macrophage production, inflammatory action that cause adipocyte death, suppression of Insulin receptor substrate 1/2 (IRS-1/2), inhibition the leptin and negatively control the obesity (Baker et al., 2011). In early stages of tumorigenesis or viral infection, the cytoprotective effect of NF-κB is beneficial because it prevents hepatocyte cell death, and thus avoiding the release of IL-1a by necrotic hepatocytes and the triggering of compensatory proliferation (Sun & Karin, 2008). In late stages of tumorigenesis, Papa et al. (2009) suggested that NF-κB supports malignancy by promoting the survival of the transformed hepatocytes against the JNK mediated injury and cell death. In non-parenchymal cells of the liver, NF-κB activation during all stages of hepato-carcinogenesis is detrimental because it provides the emerging and growing hepatoma cells with essential growth factors, allowing them to proliferate (Sakuda et al., 2002). In addition (Fig. 2) to oxidative stress, the excessive hepatocyte death in both IKKβ knockouts and IKKγ knockouts is because of prolonged activation of JNK (Sakurai et al., 2006; Sun & Karin, 2008). NF-κB is crucial for innate immune defense against microbial infection (Hayden & Ghosh, 2008; Vallabhapurapu & Karin, 2009). However, there are several regulators including bacterial infections, which show autoimmune, innate and humoral immune responses by cytokines, interleukins and other regulators. Conclusively, the roles played by NF-κB (Fig. 2) in autoimmunity, drugs which target it and complexities which need to be addressed to improve the use of NF-κB as a target has been described (O’Sullivan et al., 2007; Uwe, 2008). These facts support its master regulator activity. The immune system has evolved a variety of receptors to recognize invading microorganisms (pathogens, bacteria and viruses) which result activation of NF-κB trafficking by gene regulation reported by Rahman et al. (2011). In contrast, (Fig. 2) immune regulation by secreted pathogen derived proteins (NS3, gp350, Core) and viral coded proteins can induce TLR, TNFR and other receptors to activate MyDD88 and TAK1 (Rahman et al., 2011). It also upregulate the IκBα and control the NF-κB.
ROLE OF miRNAs AS NOVEL CLINICAL BIOLOGICAL PLAYERS IN OS
miRNAs play as mega diverse roles as tumor suppressors, oncogenes inducers or both in some cases (Davis & Hata, 2009). These qualities of miRNAs keep them as essential multipurpose prognostic and diagnostic tools in various oncogenesis including OS (Kobayashi et al., 2012). Normally miRNAs are present in various parts of the body in the form of fluids as serum, plasma, saliva, urine and milk (Chen et al., 2008; Mitchell et al., 2008; Zhou et al., 2013). Abnormal levels of miRNA can cause and are tightly linked with cancers and diseases including OS (Chen et al., 2008; Mitchell et al., 2008; Zhou et al., 2013). Studies have demonstrated diverse genetic alterations in OS cells including structural abnormalities, gain and/or loss of chromosomes, mutation in tumor suppressor genes and epigenetic modifications (Kansara & Thomas, 2007; Broadhead et al., 2011). OS is characterized by a high propensity for pulmonary metastasis. Novel therapeutic targets and approaches are required to suppress pulmonary metastasis of OS and improve prognosis which is a major cause of death (Longhi et al., 2006). NF-κB can induce the synthesis of proteins that regulate miRNAs (Hoesel & Schmid, 2013). Under normal conditions, overexpression of miRNA-194/192/215 induces the expression of p53 and p21 in OS cell lines. Furthermore, p53 dissociate from p53-specific E2 Ubiquitin protein ligase and is reported to arrest cell cycle and apoptosis to repair/damage cells (Kobayashi et al., 2012). The most important so far example is of NF-κB-dependent induction of Lin28, a protein which inhibits the processing and maturation of let-7 miRNAs. Further, members of let- 7 miRNA family are often down-regulated in cancer and which seems to act as tumor suppressor (Fig. 1). Iliopoulos et al. (2007) showed, Let-7 miRNAs target IL-6 thus a reduction of let-7 leads to higher levels of IL-6 and further activation of NF-κB generating a positive feedback loop.
Ghosh & Karin (2002) revealed, mostly in neoplasm (OS), the NF-κB pathway is switched on by a variety of cellular, developmental signals and biological factors. In contrast, a study of metastases of osteogenic sarcoma has revealed that up-regulation of GSK-3β activity is directly and jointly involved in tumor progression and clonogenicity (Yin et al., 2013). Further it has been reported that cell survival in OS in which induction of blocking of GSK-3β down-regulates the function of NF-κB pathway genes and act as anti-tumorigenic through in-vitro and mouse models (Tang et al., 2012). Therefore, therapeutic targeting of GSK-3β mediated inhibition of NF-κB pathway could be novel strategy to overcome against OS in-vitro and in-vivo. Reports by Silverman & Maniatis (2001) proved that the molecular mechanism of NF-κB signaling pathway between Drosophila and mammals is not fully understood but distinct signaling similarities are used by human and flies to activate their NF-κB trafficking. In both cases they activate TLRs to initiate intracellular signaling cascades that gave the threshold to activation of NF-κB/Rel family transcription factors. In addition, insect activate NF-κB precursor Rel by IKK and caspase-dependent endoproteolytic cleavage while mammalian NF-κB is activated by proteasome mediated destruction of IκB proteins on N-terminal inhibitory domains of NF-κB precursor (p100 and p105). Further, Perkins (2007), Karin (2009) showed, Rel family proteins exist as hetro-homodimeric proteins that are segregated in cytoplasm by interaction with various members of IκB inhibitor family proteins. Studies conducted by Perkins (2007), Karin (2009) reported, that IκB proteins are influenced by many extracellular signals, responsible for destruction of IκB inhibitor proteins by the activation of IκB kinase of IKK. However, the number of enzymes inhibit the invasion and migration of tumor propagation by several miRNAs interference which suppressed the expression of fatty acid synthase (FASN) and reduced the expression level of pAKT, NF-κB (p65), matrix metalloproteinase-2 and 9 (MMP-2 and MMP-9) proteins in OS (Liu et al., 2013). Felx et al. (2006) showed, vice-versa action that Endothelin-1 (ET-1) promotes MMP-2 and MMP-9 induction involving the transcription factor NF-κB in human osteosarcoma.
IMPORTANT SUPPRESSOR miRNAs IN OS
There are several mRNAs which can ameliorate tumor progression and metastasis by gene silencing or other ways and mechanisms (Mendell & Olson, 2012), that are required to know. In-vivo, injection of miRNA-143 down regulates proliferation and delayed tumor formation. Overexpression of miR-124 suppressed Rac1 protein expression and atenuated cell proliferation, migration, invasion and induced apotosis in MG-63 and U2OS in-vitro. Significant reduction in tumor size has been reported upon its transfer into MG- 63 and U-2 OS cell lines (Table 1). It also inhibits lung metastases in OS (Zhou et al., 2013). Tumor suppressor miRNA (miR-218) suppressed the expression level of Tcell lymphoma invasion and metastasis 1 (TIAM1), MMP- 2, MMP-9. It also inhibits OS cell migration and invasion (Jin et al., 2013). Further, the ether-à-go-go 1 (Eag1) voltage gated K+ is a central nervous system (CNS) localized channel that overexpresses in number of cancer patients it expresses in 75% solid tumor. The overexpression of miR- 34a decreased the expression level of Eag1 in in-vitro (MG-63, SaOS-2 cell lines) and in-vivo in OS (Wu et al., 2013). Many miRNAs occurred in different chromosomes in various position but most prognostic miRNAs were located at 14q32 (Kelly et al., 2013). These loci are already associated with OS which predict chemoresponses with 75 to 80% accuracy without overlaping with prognostic profile. Zhang et al. (2010) showed, miR-143 named as ‘targetome’ can inhibits many targeted mRNAs (Bcl2, DNMT3A, KRAS, and ERK5) to regulate post transcription in OS and cancer development (Table 1). Duan et al. (2011) studied, miR- 199a-3p, miR-127-3p influence the mTOR, STAT3 that arrest cell cycle (Table 1). Next, He et al. (2009) and Wu et al. (2013) showed, miR-34 family jointly with CDK6, Cyclin E2, Bcl2 possess p53 dependent apoptosis in OS (Table 1). Thayanithy et al. (2012), reported that miRNAs (miR-382, miR-134, miR-369 and miR-544) arrest cell cycle and apoptosis via down-regulating the expression of c-Myc in OS (Table 1). 3’UTR silenced of VEGF by miR- 145 in OS (Table 1) that control the tumor progression (Fan et al., 2012). The miR-218 causes lesion the OS by targeting of TIAM1, MMP-2 and MMP-9 (Table 1) (Jin et al., 2013). B-cell lymphoma-2 (Bcl2) and insulin-like growth factor 1 receptor (IGF1R) down-regulate the miR-133b Akt pathway in OS (Zhao et al., 2013). The miR-24 suppressed the expression of LPAATb in OS (Song et al., 2013). In addition, MEK and ERK Pathway regulated via downregulation of Raf by miR-16 in OS (Chen et al., 2013). Won et al. (2013) revealed, HES1 is suppressed in OS by miR199b as an inhibitor in Notch signaling cascade (Table 1).
NEOPLASIA PROMOTER miRNAs IN OS
OS are complex bone tumors with various genomic alterations (Smida et al., 2010). Polygenic spectrum of germline risk factors pointing towards specific pathways as drivers of disease; it implicates 33 inherited risk loci in heritable dog osteosarcoma, including regulatory variants near CDKN2A/B (Karlsson et al., 2013). These alterations affect the expression and function of several genes by overexpressed miRNAs in OS patients (Namløs et al., 2012). Genomic alterations are also associated with several transcription factor (TF), co-regulate networks in OS cell proliferation (Poos et al., 2013). Some clinically candidate miRNAs (miR-196a and miR-196b) have been demonstrated to play a crucial role in normal cell differentiation, proliferation and in tumorgenesis of various cancer types (Table 1) including OS cell lines relative to normal bone (Namløs et al., 2012; Liu et al., 2013; Zhang et al., 2014). Another miR-33a is up-regulated in chemoresistant OS (Fig. 1). The level of miR-33a is negatively correlated with the TWIST protein level and the tumor necrosis rate in OS (Zhou et al., 2014). In addition, miR-33a promotes OS cell resistance to cisplatin by down-regulating TWIST. Actually, TWIST is a pivotal negative regulator of OS chemoresistance (Table 2). Several miRNAs are key of modulator specifically, 14q32 miRNAs are up-regulated upon osteoblast maturation while overexpression (Exogenous) of 14q32 miRNAs leads to reduction in cMYC protein by blocking miR-17-92 or cMYC depletion which induces apoptosis in OS (Thayanithy et al., 2012; Shimada, 2013). Invasiveness induced through miR-362 by induced nuclear translocation of p65 and inversely regulates the CYLD expression by binding its 3’untranslated region in Gastric cancer (Fig. 1) including OS (Xia et al., 2014). Dysfunctional NF-κB activity contributes to various human conditions, including autoimmune, inflammatory disorders and cancer by miR- 125a and miR-125b (Kim et al., 2012; Tan et al., 2012). It directly Inhibit tumor necrosis factor alpha-induced protein 3 (TNFAIP3) expression, down-regulate the p38α via UV that elevates NF-κB Activity in diffuse large B-cell lymphoma (DLBCL) and OS (Fig. 1). Won et al. (2013) showed, the expression of 10 miRNAs (HS-169, HS-29, miR-10b, miR-150, miR-199b-5p, miR-223, miR-338-3p, miR-451, miR891a and solexa-578-1915) has been reported to increase 10-fold in OS as compared with normal controls by microarray analysis (Table 2). Ziyan et al. (2011) reported, potential regulation of RECK by miR-21; it is over expressed in OS.
A BRIDGING AVENUE TO AMELIORATE ONCOGENESIS THROUGH miRNAs AND NF-κB
The nuclear factor-KappaB (NF-κB) pathway is conserved from fruit flies to humans and is a key mediator of inflammatory signaling (Minakhina & Steward, 2006; Olarerin-George et al., 2013). Aberrant regulation of NF-κB is reported to be associated with autoimmune diseases, chronic inflammation and cancers (Silverman & Maniatis, 2001). It makes the NF-κB pathway an attractive therapeutic target (Olarerin-George et al., 2013). NF-κB is a transcriptional regulator consisting of reticuloendotheliosis (Rel) protein dimers that bind a DNA sequence motif known as κB site (Karin, 2009). NF-κB activation is a result of underlying inflammation or the consequence of formation of an inflammatory microenvironment during malignant progression (Perkins, 2007; Karin, 2009). It has ability to up-regulate the expression of tumor promoting cytokines, such as IL-6 or STAT3 and survival genes, such as Bcl-XL (Perkins, 2007; Karin, 2009). The basic-helix-loop-helix (b-HLH) factor Twist-1 is reported as a new mediator of the protective function of NF-κB and it blocks PCD induced by chemotherapeutic drugs and TNF-alpha in NF-κB-deficient cells (Pham et al., 2007). It is also essential to counter this PCD in cancer cells via dephosphorylation of BCl-2. Next, Tan et al. (2012) showed, activation of NF-κB by UV is required for miR-125b induction in colon cancer and OS. Deregulated miR-143 in cancers including OS directly target BCl2 and anti-apoptotic molecule and regulate NF- κB (Zhang et al., 2010). While, Chen et al. (2013) demonstrated the miR-16 inhibits the proliferation and metastasis of OS by targeting IGF1R and suppression of Raf-MEK1/2, ERK1/2 in nude mouse (Table 1). Geng et al. (2014) reported that Rac1 targeted by miR-124 that attenuated the cell proliferation, invasion and metastasis in MG-63 U2OS cell lines. Similarly, miR-26b (Fig. 1) inhibited the expression of TAK1 and TAB3 by binding to their 3’-untranslated region in hapatocarcinoma cells (Zhao et al., 2014). Let-7 and miR-34 family directly suppress the IL-6, STAT3 and MMP-9 (Table 1) which down-regulate the Rel family subunits and palliate from carcinogenesis (He et al., 2009; Iliopoulos et al., 2009; Zhou et al., 2013). Although, these miRNAs can regulate not only NF-κB bio-molecular mechanism but also involve in various type of signaling pathways in neoplasia (Davis & Hata, 2009). On the other hand, few cancer inducer miRNAs (miR-362, miR-125b) are reported to up-regulate the Rel protein expression in invasived and metastasized tumors (Fig. 1 & Table 2) (Kim et al., 2012; Tan et al., 2012; Xia et al., 2014). Moreover, miR-33a, overexpression causes drug resistance in OS by targeting TWIST (Zhou et al., 2014). Apart from these miRNAs there are several another which are yet to be known. Notably, the targeted genes mediated activation of miRNAs represses the tumor proliferation and metastasization by novel approach to link between miRNAs and NF-κB as a bridge model in cancer biology. Moreover, we studied the inverse correlation or vice-versa of miRNAs and several signaling key molecules including Rel family (NF-κB) expression in a panel of neoplasm (OS) patientderived xenografts as well as in-vitro model. Finally, we show that this bridge model can be a step ahead to overcome the issue of drug resistance and it also suggests that miRNAs combine with NF-κB could be involved in resistance to drug sensitivity of cancer stem cells and its inhibition may be exploited in target therapy protocols.
CONCLUDING REMARKS
Although many phosphorylation sites on NF-κB proteins have been characterized, it is still unclear how these phosphorylations regulate the ability of such proteins to induce or to repress defined target genes. Self-renewing solid and soft cancer stem cells are the subject of intensive research, as these are the key factors responsible for perpetuating tumor existence, for treatment of resistance and relapse. Tumor-promoting inflammation has finally been recognized as one of the hallmarks of cancer. Carcinogenesis is a multistage process in the classical sequence of chemical carcinogenesis; tumor initiation is followed by tumor promotion. Human epidemiology and animal model studies indicate that chronic, smoldering inflammation may be a far more widespread ground for cancer development than the previous thoughts. At the same time, NF-κB activation, acting as one of the pillars of inflammation, may have a promoting role in most cancers. Twenty-five years after the discovery of NF-κB, meager information is available about its signaling and transcriptional targets. Taming of NF-κB activity still remains an important challenge for modern cancer biology. Appearance of IKKε to act as an oncogenic protein through NF-κB, yet is totally unclear whether and how this kinase promotes breast cancer stem cell expansion. Answers to all such thoughts might come by the knock-in experiments in which the combined approach of “bridge between NF-κB and miRNAs (BBNM)” is adopted. Use of mice for in-vivo phenotypic analysis may provide a powerful biological model to address the regulation of NF-κB protein activities by phosphorylation. Moreover, now it seems that aberrant regulation of NF-κB could also underlie different type of cancers along with autoimmune, metabolic and neuronal diseases. Recent development in research to find the mechanism of OS progression is not adequate although mixed-model genome wide association study (GWAS) of OS has demonstrated regions associated with tumor progression in dog breeds (Karlsson et al., 2013). This approach can be used to study the cancer progression and metastasis. Despite of the significant progress in the elucidation of the roles played by NF-κB in cancers including OS cancer stem cell expansion, some issues remain to be experimentally addressed. Candidate genes known to regulate embryonic stem cells and to be specifically induced through IKKα activation remain to be identified. Although tremendous advances have been made in characterizing the role of the IKK complex and NF-κB subunits, as well as upstream modulators such as TRAFs and RIPs, but still facts about how these signaling modules affect non-NF-κB pathways remain to be learnt. However, clear genetic proof of crosstalk between pathways is difficult to obtain as it is necessarily obfuscated by deficits in the main pathway in which a given signaling component functions. The same holds true for interpretation of the biological outcomes of deleting signaling molecules that may bridge NF-κB to other pathways and several miRNAs which are responsible for key regulation of all biological processes. In this study, we have tried to correlate NF-κB and their all components with many miRNAs as “Bridge Model” to make novel and innovative approach to ameliorate the inflammatory, metabolic, neuronal, bacterial/ viral transmitted diseases and cancer. The application of NF-κB inhibitors in combination with miRNAs is expected to result in a more efficient killing of the cancer stem cells and a slower or less likely recurrence of cancer. Another important aspect is that inhibition of NF-κB might target cancer not only directly by blocking anti-apoptosis mechanisms of malignant cells, but also indirectly by shifting macrophages from the tumor-tolerating M2-polarization stage to the tumor attacking M1-stage by neoplasia suppressor miRNAs.