
INTRODUCTION
Organoid mimics in vivo organs both structurally and functionally, which enables researchers to gain a deeper understanding of human organ development, physiology, and pathology. Two methods for generating organoids are currently used. The first involves using tissue stem/progenitor cells. In 2009, Hans Clevers and his team showed that leucine-rich repeat-containing G protein-coupled receptor-5 (LGR-5) positive stem cells could generate three-dimensional intestinal organoids (Sato et al., 2009). Since then, many other organoids, such as those of the liver, kidney, pancreas, stomach, uterus, prostate, and mammary gland, have been generated using stem cells from their own respective tissues (Barker et al., 2010; Karthaus et al., 2014; Boj et al., 2015; Hu et al., 2018; Sachs et al., 2018; Fitzgerald et al., 2019; Schutgens et al., 2019). The second method involves using pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), which have the capacity for self-renewal and differentiation (Fatehullah et al., 2016). PSCs can differentiate into three germ layer cells using signaling pathways such as wnt, retinoic acid, fibroblast growth factor, bone morphogenic protein, and transforming growth factor (Clevers, 2016). Recently, organoids of various organs, such as the intestine, liver, lung, thyroid, pancreas, brain, retina, and kidney, have been generated using PSCs (Eiraku et al., 2008; Kurmann et al., 2015; Ogawa et al., 2015; Takasato et al., 2015; Chen et al., 2017; Hohwieler et al., 2017; Tsai et al., 2017; Capowski et al., 2019).
Kidney is complex organ that is compose of various different types of cells and it is responsible for many functions, such as filtering and removing the waste products form the body, removing drugs from the body, balancing the fluid in the body, releasing hormones that regulate blood pressure, and control the production red blood cells. The basic functional unit of kidney is called nephron consists of glomerulus, a complex of blood vessel capillaries and podocytes responsible for the filtration of blood, and multiple segment of tubule epithelium responsible for reabsorption and hormone secretion. Loss of functional nephrons and the development of tubulointerstitial fibrosis contribute to the progression of chronic kidney disease, and it affects around 15% of the population and it ultimately leads to end-stage kidney disease (ESKD; Coresh et al., 2007; Eneanya et al., 2016). The patients of ESKD require renal replacement therapies, such as hemodialysis, associated with high morbidity and mortality. Moreover, in vivo mimic disease models for biomarker identification and development of therapeutic approaches for most kidney diseases are limited. A better understanding the basic mechanism of ESKD will be useful to develop novel therapeutic approaches and prevent the disease progression. In general, the causes of kidney failure could be divided into genetic and non-genetic causes. Several differentiation protocols for kidney organoids have been reported from PSCs, and some of these reports mimicked disease models using kidney organoid by genetic mutant line generation. Here, in this review, we discuss genetic kidney disease modeling using PSCs and the kidney cancer organoids using patient-derived samples.
KIDNEY ORGANOID FOR KIDNEY DISEASE MODELS
Human PSCs are highly adaptable to genetic manipulation, they can cause genetic abnormalities associated with hereditary kidney disease. Polycystic kidney disease (PKD) has been most frequently studied using PSCs derived kidney organoid. Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease, and the main feature of ADPKD are multiple renal cysts that eventually cause renal failure, often accompanied by liver, pancreatic cysts, and cerebral aneurysms (Fig. 1A; Torres et al., 2007). PKD1 and PKD2 are the main causative genes for ADPKD, the former accounting for about 85% of cases. CRISPR-Cas9 PKD1-edited biallelic mutant ESCs derived organoid showed formation of cyst-like structures in the proximal tubule of organoid (Freedman et al., 2015; Cruz et al., 2017) and the kidney organoid from CRISPR-Cas9 PKD1 edited human iPSCs cell line exhibited cysts formation in both nephron progenitor and UB/collecting duct tubules in organoid after cyclic adenosin monophosphate (cAMP) stimulation (Kuraoka et al., 2020; Shimizu et al., 2020). Biallelic gene-edited mutant organoids displayed cyst formation following cAMP activation, suggesting that cAMP signaling is important in PKD cystogenesis in cellular models as well as in vivo (Harris & Torres, 2014). Autosomal recessive PKD is characterized by enlarged kidneys with 2 waves of cytogenesis. In ARPKD kidney, proximal tubule cysts are prominent in fetal kidney, and cyst formation dominates in the collecting duct after birth (Nakanishi et al., 2000). Low and colleagues reported the kidney organoid from patient derived biallelic PKHD1 mutant hiPSCs showed cysts formation in the proximal tubules following cAMP stimulation, and CRISPR-Cas9 corrected isogenic lines produce organoids with ameliorated cytogenesis (Low et al., 2019).
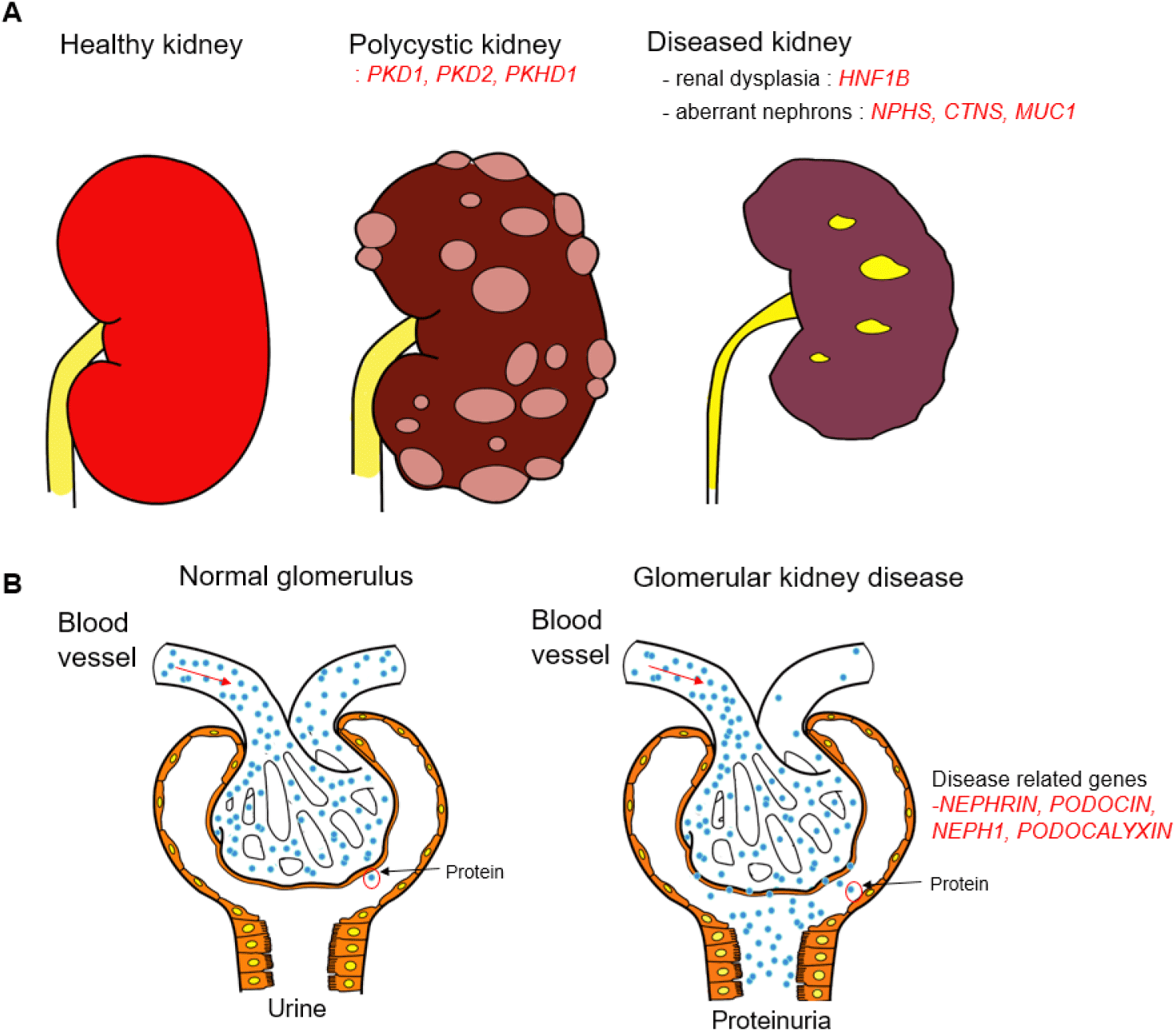
HNF1B is expressed in developing mouse ureters and collecting ducts, and it is expressed in proximal and distal tubules after birth (Coffinier et al., 1999). HNF1B is expressed in both maturing human collecting ducts and nephrons. Hnf1b deletion in mouse collecting ducts causes cysts and upregulates transcription of uromodulin (Umod), Pkhd1, and Pkd2 (Gresh et al., 2004), and similar results are also observed in human kidneys such as fetal bilateral hyperechogenic kidneys, multi-cystic dysplastic kidneys, and hyperuricemic nephropathy (Fig. 1A; Bingham et al., 2003; Ulinski et al., 2006; Decramer et al., 2007). CRISPR-Cas9 HNF1B edited biallelic or heterozygous mutant iPSCs derived kidney organoid exhibited the reduced proximal tubule, thick ascending limb markers and reduced UB-derived tubule branching (Przepiorski et al., 2018; Mae et al., 2020). hPSCs derived kidney organoids have also been used to model for genetic tubulopathies, such as cystinosin (CTNS) and Mucin-1 (MUC1) mutation for nephropathic cystinosis and medullary cystic kidney disease, respectively (Fig. 1A). In the absence of CTNS, cystine accumulates within the lysosome, causing lysosomal dysfunction. Nephropathy is the most severe form of cystinosis and initially involves the inability of the renal proximal tubules to reabsorb essential metabolites from the urine (Gahl et al., 2002). Hollywood and colleagues generated CTNS mutant iPSCs and showed that kidney organoid from these iPSCs exhibit elevated cystine levels, enlarged lysosomes, increased apoptosis, and defective basal autophagy, and they demonstrated that combined treatment with cysteamine and everolimus, an mTOR pathway inhibitor, rescued the disease phenotype in the mutant hiPSCs derived organoid (Hollywood et al., 2020).
Autosomal dominant tubulo-interstitial kidney disease-mucin1 is caused by a frame-shifting mutation in the GC-rich variable number of tandem repeats region of the MUC1 gene (Kirby et al., 2013). In the kidney, MUC1 localizes to distal convoluted tubules and collecting duct, and after ischemia induction, the protein may be induced in the proximal tubule (Al-Bataineh et al., 2016; Gibier et al., 2017). Missense MUC1 mutation generates a protein accumulation with in the epithelial cells in the patient derived hiPSC kidney organoids leading to damages the tubules by accumulating in transmembrane emp24 domain-containing protein (TMED9) enriched vesicles, and treatment with the small molecule BRD4780 binding cargo receptor TMED9 significantly reduced levels of misfolded MUC1 (Dvela-Levitt et al., 2019).
Our understanding of glomerular nephropathy is hampered by the limited proliferative nature and architecturally complex structure of the primary podocytes (Lasagni et al., 2013). In the glomerulus, podocytes play an important role in the filtration process and have multiple cytoplasmic protrusions called foot processes. The podocyte foot processes form a specialized cell–cell contact, the slit diaphragm (SD) to prevent leakage of high molecular weight serum proteins into the urine (Schell et al., 2014; Fig. 1B). The main components of SD are NEPHRIN, PODOCIN, and NEPH1. NEPHRIN and HEPH1 are transmembrane proteins that intercalate with proteins from adjacent cells to form a molecular mesh, SD. PODOCIN is thought to stabilize SD by binding to the cytoplasmic region of NEPHRIN (Sharmin et al., 2016). Thus, mutations in these genes cause proteinuria in humans and/or mice. NPHS1 mutated hPSCs derived kidney organoid models reduced levels of nephrin and podocin SD proteins and NPHS1 protein does not localize to the surface of podocyte-like cells (Tanigawa et al., 2018). Mutation in NPHS1 showed abnormality in the SD formation. Podocyte derived from NPHS1 mutant patient iPSCs showed significantly reduced cell surface localization of NEPHRIN despite having normal foot process (Tanigawa et al., 2018). Other study demonstrated that mutation of NPHS1 showed reduced levels of NEPHRIN and PODOCIN SD proteins and large hypertrophied podocyte bodies in the iPSCs derived kidney organoid (Hale et al., 2018). These models are useful for studying of congenital nephrotic syndrome.
Homozygous loss of PODOCALYXIN (PODXL) leads to perinatal retention of junctional complexes between immature podocytes, a walling off the urinary space, renal failure and ultimately, perinatal death (Doyonnas et al., 2001; Kang et al., 2017). Therefore, PODXL mutant hPSCs derive kidney organoid is useful for studying human glomerular development. CRISPR-Cas9 edited biallelic mutant hESCs derived kidney organoid exhibited a lack of microvilli on apical and lateral podocyte cell membrane and reduced lateral spacing (Kim et al., 2017). Moreover, this model showed defective junctional organization and decreased gaps between adjacent podocytes similar to that observed in Podxl null mice (Freedman et al., 2015). This kidney organoids suggest the potential of kidney organoids as an effective model for studying pathological mechanisms for glomerular kidney disease.
Renal cell carcinomas (RCCs) refer to a group of cancers that can be identified by their distinct genetic mutations. There are about 372,000 new cases of kidney cancer diagnosed worldwide each year, with approximately 166,000 deaths reported in 2019. The three primary subtypes of RCC are clear cell RCC (ccRCC), papillary RCC (pRCC), and chromophobe RCC (chRCC), which account for 75%, 15%, and 5% of RCCs, respectively (Creighton et al., 2013; Davis et al., 2014; Marston Linehan et al., 2016; Hsieh et al., 2017). ccRCC is the most common subtype of RCC and is characterized by a loss of function mutation in the von Hippel-Lindau gene, leading to the accumulation of hypoxia-inducible factor (HIF) and subsequent activation of angiogenesis and cell proliferation pathways. pRCC is characterized by a mutation in the MET gene or its downstream signaling pathways, leading to aberrant cell proliferation and invasion. chRCC is characterized by alterations in mitochondrial genes and metabolic pathways. Inherited mutations can increase the risk of developing RCC and pose a significant challenge, as there is currently a lack of three-dimensional in vitro models for studying cancer progression.
Recent advancements in the field of renal cancer research have led to the development and characterization of tumoroids derived from renal cancer cells. These tumoroids have proven to be effective in preserving critical genetic and phenotypic features of the original tumor tissues. ccRCC-derived tumoroids have exhibited the presence of both epithelial and mesenchymal cells, expressing specific markers associated with renal cancer, such as HIF1α. Importantly, these tumoroids have demonstrated the ability to proliferate even after being transplanted into xenograft models (Grassi et al., 2019). In a separate study conducted a biobank was established using various childhood kidney cancers, including Wilms tumors, RCC, and malignant rhabdoid tumors of the kidney (MRTK). The tumoroids derived from these samples exhibited tri-phasic histology, consisting of epithelial, stromal, and blastema components. Notably, the MRTK tumoroids represented a significant breakthrough as they were the first cancer organoids capable of long-term in vitro expansion for non-epithelial origin tumors (Calandrini et al., 2020). Huang group developed the kidney organoid culture system utilized to generate 33 kidney cancer organoid lines from common subtypes of kidney cancer, including ccRCC, pRCC, and chRCC. These RCC organoids retained the histological structures, mutational characteristics, and transcriptional profiles of the original tumor tissues. Furthermore, single-cell RNA sequencing demonstrated inter- and intra-tumoral heterogeneity in RCC organoids. RCC organoids also enabled in vitro drug screening and offered a means of evaluating the effectiveness of chimeric antigen receptor T cells (Li et al., 2022).
There is a study to report the in vitro model of c-met-mutated hereditary kidney cancer using hiPSCs derived from a patient with type 1 pRCC. The hiPSCs were able to differentiate into 3D kidney organoids that exhibited features of glomeruli, proximal tubules, and expressed markers of pRCC, renal progenitors, and endothelial cells. These organoids were then transplanted under the kidney capsule of NSG mice, where they formed larger tumors compared to the controls. The researchers also found that the gene expression signature of these organoids was highly associated with the expression pattern found in a large cohort of pRCC patient samples. They identified 11 common genes, including BHLHE40 and KDM4C, which are factors involved in pRCC pathogenesis. This study provides a promising in vitro model for studying c-met-mutated hereditary kidney cancer and may lead to better understanding of the disease and development of targeted therapies (Hwang et al., 2019). In addition, Schutgens et al. (2019) used adult stem cells isolated from the urine of patients with various kidney diseases to generate kidney tubuloids. These tubuloids were used to study the effects of BK virus on nephrotic injury, investigate the effects of agents on cystic fibrosis phenotypes, and establish tubuloids from Wilms tumor tissue to study the disease’s pathogenesis (Schutgens et al., 2019). Despite these promising developments, it is important to note that research utilizing renal cancer-derived tumoroids is still in its early stages. Further improvements in the methodology are necessary to enhance the potential applications of these tumoroids in downstream studies and clinical applications. Nevertheless, a lot of studies are currently underway to better understand the molecular mechanisms of RCC and to develop more effective treatments. The use of kidney organoids derived from patient-specific tissue or iPSCs holds promise for personalized drug screening and precision medicine approaches in the treatment of RCC development.
CONCLUSION
The development of kidney organoids using human adult or PSCs holds great significance in understanding kidney development and modeling kidney diseases. Kidney organoids allow for personalized studies of genetic kidney diseases and drug screening on a human-derived platform. Although they have tremendous promise, kidney organoids currently have limitations in terms of maturity and inability to represent all types of kidney cell. To develop the in vivo mimic human kidney and related diseases, we need to mature them by recapitulating developing kidney interactions, especially between the UB and MM, and focus on the development of vascularized organoids, given the intimate relationship between the kidney and vasculature. Kidney organoids have the potential to be used for genetic studies of adult and fetal kidney disease, drug screening, and personalized medicine development. The progress in kidney organoid technology has the potential to expedite the development of more precise and efficient kidney disease treatments.