
Lung cancer is the leading cause of cancer deaths among both men and women. The American Cancer Society indicates that there were more than 130,000 deaths from lung cancer in 2022 in the United States alone. Small-cell lung cancer (SCLC) accounts for approximately 10%–15% of all lung cancers and is a high-grade neuroendocrine neoplasm that is distinct from the rare subtypes of large cell neuroendocrine carcinoma and neuroendocrine carcinoid (Lewis et al., 2014; Park et al., 2016; Torre et al., 2016). It is an exceptionally lethal type of lung cancer with a 5-year survival rate of 7%, significantly worse than that of NSCLC (26%) (Byers & Rudin, 2015; Rudin et al., 2021). The high mortality of SCLC patients is mainly due to the highly metastatic nature of the cancer and the invariable resistance to current therapies that have remained largely unchanged for the past 30 years. Despite the recent success of tobacco control policy and smoking cessation campaigns in reducing the disease incidence, the number of the patients will remain high for decades to come, not only because former smokers would still develop the cancer after a long latency but also because approximately 20% of the adult population in the US continues to smoke. While the long tumor latency and the known risk population of SCLC rationalize chemoprevention, the paucity of molecular targets for chemoprevention stems from the poor understanding of the SCLC progression beyond the role of RB and P53 mutations crucial for tumor development. The conventional chemotherapy of cisplatin and etoposide has not significantly improved overall survival in SCLC patients and the development of targeted therapies has been challenging due to the paucity of clinically viable targets. SCLC is mostly driven by loss of tumor suppressors, including inactivation of RB and P53 and frequent loss of other tumor suppressors such as the RB family members RBL1 and RBL2 (coded by the RBL1 and RBL2 and also known as p107 and p130, respectively) (Peifer et al., 2012; Rudin et al., 2012; George et al., 2015; Augert et al., 2017). A better way to identify the mechanism underlying the development of resistance may lead to the development of a novel therapeutic strategy. In addition, an advanced understanding of the mechanisms of tumor initiation and early-stage progression may attribute to the development of novel means for early detection and prevention.
Functional characterization of the relevant cancer genes has been of prime interest but challenging due to lack of systematic and informed ways of defining oncogenic drivers. Particularly, defining gene function during early cancer progression is extremely difficult in most cancer types, because pre-cancerous lesion is often not accessible for functional study and cell lines derived from full-blown tumors may not be an ideal model (Weinstein et al., 2013). Genetically-engineered mouse model (GEMM) has provided a unique opportunity to understand the mechanisms of cancer progression and identify potential therapeutic targets (Friedel et al., 2007; Hansen et al., 2008; Walrath et al., 2010). However, the GEMM usually requires combining multiple alleles through a new gene, limiting characterization of a large number of mutations and also interactions among heterogeneous genetic factors driving tumor development. Recent studies present a 2-step approach for testing the disease relevance of uncharacterized somatic alterations found in SCLC. First, in an in vitro model, we induce candidate mutations in pre-cancerous cells (preSC) derived from the GEMM and characterize cellular phenotypes for transformation (Kim et al., 2016; Jia et al., 2018; Kim et al., 2020; Kim et al., 2022a). Second, we suggest in an in vivo model where we induce the in vitro prioritized mutations in the GEMM, using adenoviral delivery of clustered regularly interspaced short palindromic repeats (CRISPR)-Cre hybrid system (Kim et al., 2022b). The successful outcome of the models will lead to identification of key drivers of SCLC, paving the way for future experiments aimed at novel strategies for prevention and treatment, and also introduce new approaches that can be readily applied to study a wide range of cancer types that face similar research challenge.
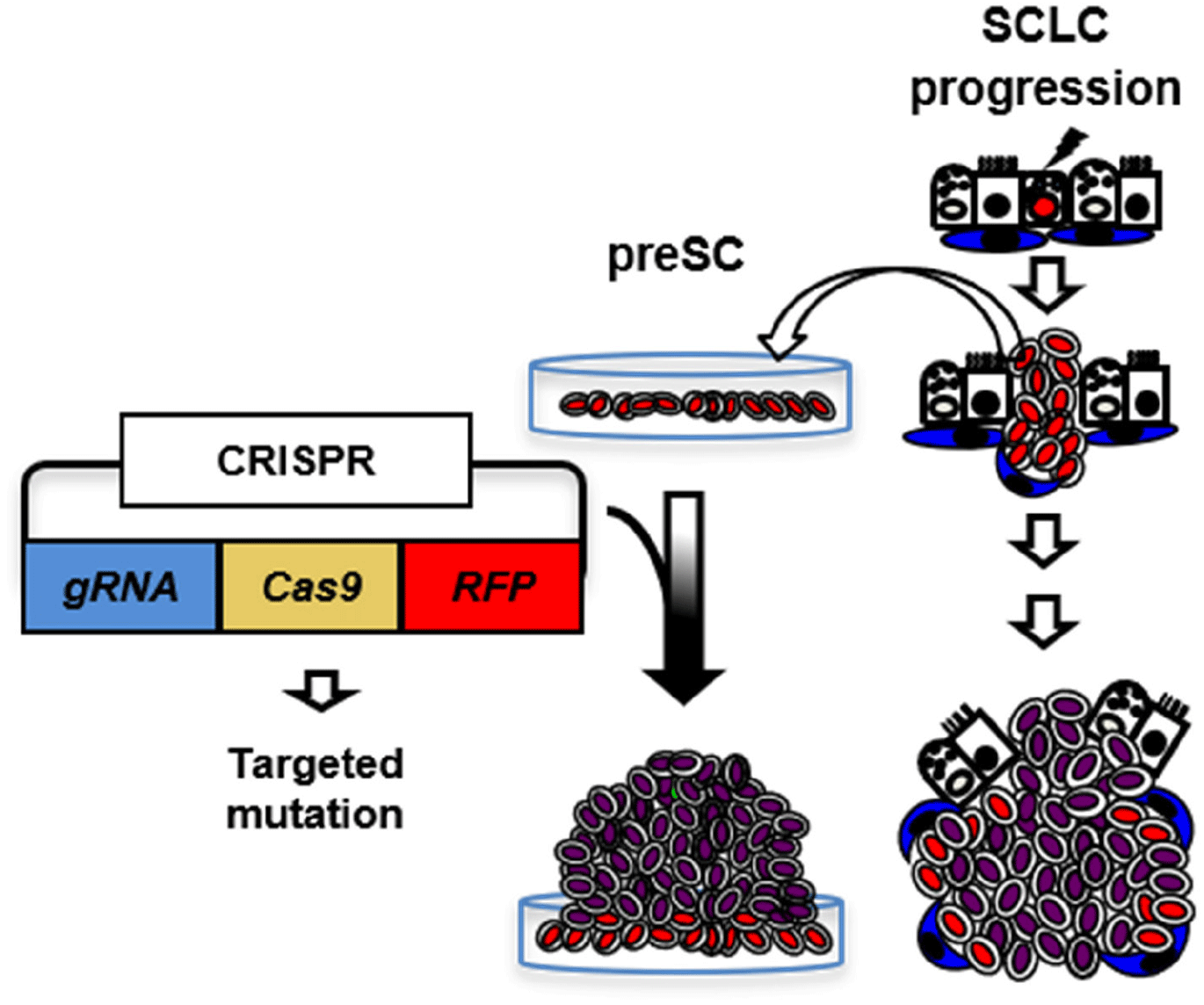
ISOLATION OF PRE-CANCEROUS PRECURSOR OF SMALL-CELL LUNG CANCER (SCLC)
GEMM was developed based on the frequent RB and P53 mutations (≥90%) in human SCLC (Peifer et al., 2012; Rudin et al., 2012; George et al., 2015; Augert et al., 2017). Infection of lung epithelial cells of mice carrying floxed alleles of Rb and p53 (Rblox/lox; p53lox/lox) with adenovirus expressing Cre-recombinase (Ad-Cre) results in loss of function, and the ‘Rb/p53’ mutant lung develops tumors that closely resemble histopathological features similar to those of human SCLC, including neuroendocrine differentiation, genome-wide gene expression, and metastasis patterns. Recently, we developed a variant of the GEMM bearing additional loss of Rbl2 (a Rb family member, also known as p130). This ‘Rb/p53/Rbl2’ model develops more SCLC tumors with shorter latency compared to the ‘Rb/p53’ model while the two models are almost identical at the molecular level and both resembles human SCLC (Schaffer et al., 2010). Recent validation of lung neuroendocrine cells (LNECs) as SCLC cell-of-origin enables the characterization of mutant cells at any stage of tumor progression using the mouse model combined with neuroendocrine-lineage specific tools (Kim et al., 2016). Using a neuroendocrine-specific reporter mouse strain (Chga-GFP) that expresses GFP under control of the transcription regulatory elements of Chromogranin A gene (ChgA), a pan-neuroendocrine marker (Oberg et al., 2015; Balanis et al., 2019) specifically labeled LNECs (Fig. 1). To facilitate characterization of mutant cells at any stage of tumor progression, we crossed the Chga-GFP strain with the GEMM of SCLC to generate ChgA-GFP/Rb/p53/Rbl2 and isolated GFP-positive cells from the lungs one month after Ad-Cre infection, when the lungs did not show macroscopic lesions, or full-blown tumors. Notably, the cells from early stage grew in monolayers, whereas the tumor cells isolated from fully grown tumors formed spheres or aggregates and grew in suspension or loosely attached to culture dish, similarly to human SCLC cells. These results indicate that these cells are immortalized due to the deletions of Rb and p53 (a common cause of cell immortalization), yet not tumorigenic because these cells, unlike SCLC cells, lack oncogenic factors, such as L-Myc, but maintain a high level of E-cadherin, while both cell types express Chga (Cardnell et al., 2017; Mollaoglu et al., 2017; Chalishazar et al., 2019; Gay et al., 2021). Therefore, we postulate that these cells are pre-cancerous precursors of SCLC tumor (herein ‘preSC’). This model was developed based on the pre-malignancy of preSC derived from the GEMM of SCLC using a lineage-labeling tool. It serves as an efficient system to characterize candidate drivers for their abilities to transform preSC to tumorigenic cells in culture and allograft experiment with immune competent mice. In addition, comparative gene expression profiling of preSC and tumor cells, so called “apple-to-apple approach”, enables identification of genes and pathways that are directly related to the pathogenesis of the cancer. Given a turnaround time of 2 months on average to test a gene as well as the capacity of simultaneously testing multiple genes, this approach should enable functional interrogation of a large number of genes, increasing the chance of finding key oncogenic drivers.
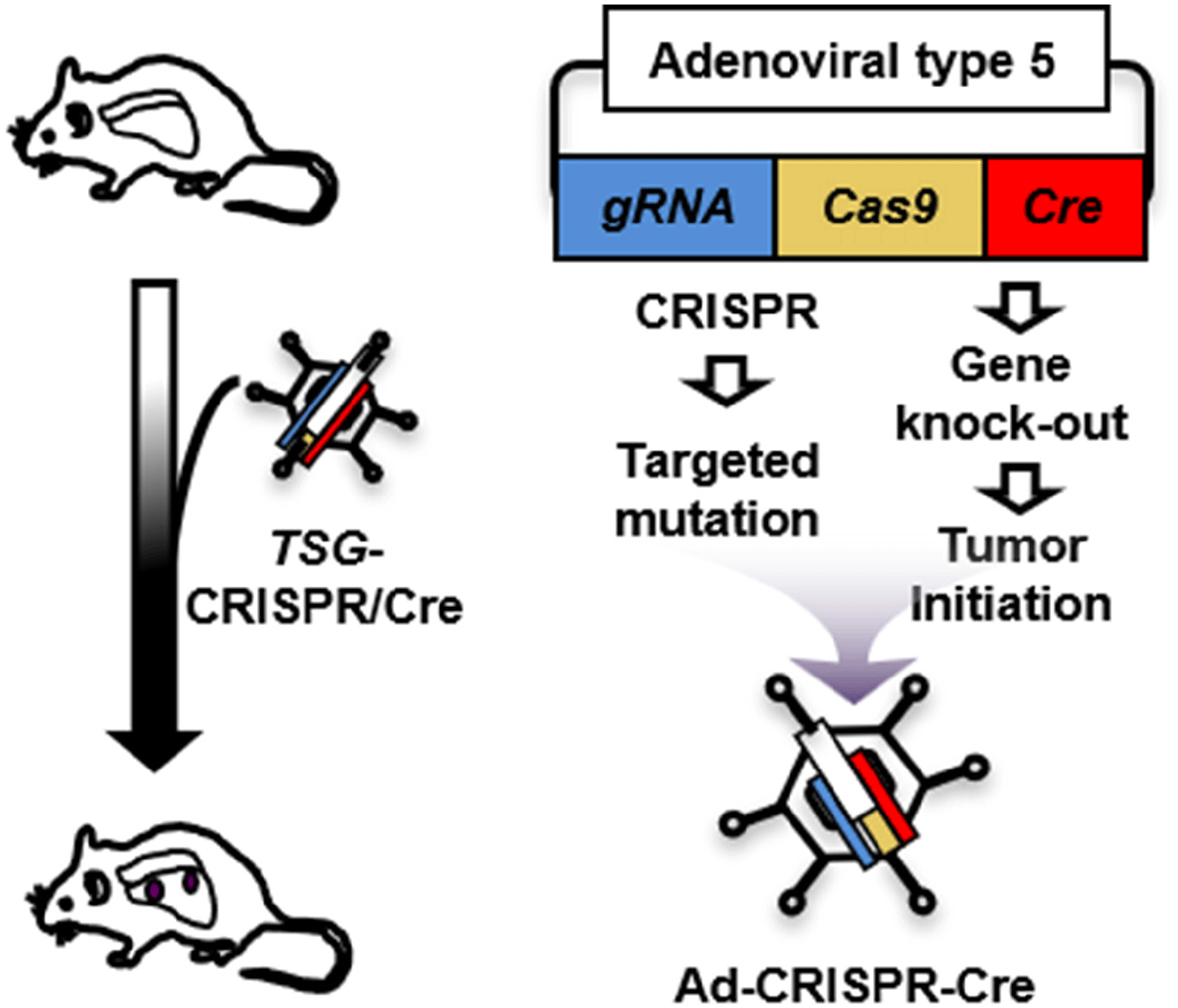
IN VIVO VALIDATION SMALL-CELL LUNG CANCER (SCLC) MUTATIONS USING AN ADENOVIRAL CLUSTERED REGULARLY INTERSPACED SHORT PALINDROMIC REPEATS (CRISPR)-CRE HYBRID SYSTEM
CRISPR, a microbial nuclease system involved in defense against invading phages, has been modified as a molecular tool for genome engineering at the cellular or organic level, simply using guide RNA and CRISPR-associated nuclease (Cas) (Cong et al., 2013; Mali et al., 2013). Briefly, a guide RNA with a complementary sequence recruits Cas to a target genomic sequence. Cas then causes a double-stranded break that triggers the error-prone repair mechanisms of non-homologous end-joining. The repair process often results in frame shift, deletion, or insertion mutations.
Unlike modeling gene amplification via lentiviral transduction, recapitulation of somatic mutation in vivo has difficult, which limits characterization of tumor suppressor genes (TSGs). Here we propose to develop a strategy for efficient functional validation of oncogenic drivers in vivo, by introducing candidate mutations in the GEMM using CRISPR-mediated genome editing, and characterize tumors whether they resemble autochthonous tumors of the mouse model as well as human tumors. We engineered a novel CRISPR-Cre hybrid vector by genetically fusing the CRISPR cassette to Cre through a self-cleaving peptide T2A derived from Thosea asigna virus (Fig. 2) (Kim et al., 2022b). The vector expresses guide RNA and Cas-2A-Cre fusion protein in the cells. Self-cleavage of 2A separate Cas and Cre that result in CRISPR-mediated mutation and deletion of floxed Rb and p53 alleles, respectively, in the same cell. For in vivo use, we incorporated this hybrid system in an adenoviral vector (pAd/PL-DEST) using Gateway cloning kit from Life Technologies (Russell et al., 2000). We validated the adenoviral vector using a mouse Cre-reporter cell line that expresses YFP upon Cre-mediated deletion of lox-stop-lox cassette that usually blocks the gene expression. We confirmed the mutations in the target region of the genome of the infected cells while Ad-Cre control did not induce mutation (Kim et al., 2022b). This adenoviral vector will produce the serotype 5 virus (Ad5), the same type as Ad-Cre that we routinely use to infect the lungs of GEMM. High-titer virus particles are produced and accurately titrated by Vector Development Lab at Baylor College of Medicine, the source of Ad-Cre for many lung cancer researches. CRISPR approaches have just merged for the models of liver and non-small cell lung cancers (Xue et al., 2014; Yin et al., 2014; Rogers et al., 2018; Ng et al., 2020). Despite their significance, these approaches still require a combination of a GEMM and a CRISPR system to study many cancer types, including SCLC, that are thought to be initiated by mutation of multiple alleles. Our CRISPR-Cre hybrid system is capable of initiating tumor and altering specific genomic sequence in the same cell. Adenoviral delivery of this system enables functional characterization of candidate gene in vivo, overcoming challenges that traditional mouse genetics approaches face to study numerous genetic factors. This combination of the CRISPR-Cre hybrid system with the GEMM is tractable and robust in characterizing a large number of mutations and genes. Our new model and approach will accelerate efforts to understand the biology of SCLC and also other cancer models that are accessible to virus-mediated gene delivery.
CONCLUSION
We expect increased tumorigenesis in the mice of the experimental group. If the mice develop larger tumors than the controls. This would mean that the introduced mutations caused increased proliferation rate and/or earlier tumor initiation. This can be differentiated by monitoring the mouse lungs using bioimaging instruments over time. In case of accelerated progression, the tumors in the experimental group would display features of higher-grades and be more metastatic compared to those in the controls. If tumor number also increases, that would mean that the genes similar to Ep300 normally suppress tumor initiation as well as progression. However, if the tumors of the experimental group is not significantly different from those of controls but have intended mutations, this will indicate that the mutations are not sufficient to drive in vivo tumor progression.
Defining patient mutations will advance the field of SCLC, paving the way to mechanistic elucidation and integration of oncogenic pathways and leading to development of a targeted therapy or optimal drug selections for treatment. Furthermore, the new technique, integrating tractable models of tumor progression with high-resolution genomic data, will impact current efforts aimed at discovery of ‘cancer drivers’.