
INTRODUCTION
Scientific and public interest in the impact of microbiomes on human health has increased dramatically over the last decades. Microbial communities in the human body, consisting of bacteria, viruses, and fungi, are involved in the regulation of immune responses and have been linked to a variety of diseases (Lin et al., 2014; Palm et al., 2015; Lahiri et al., 2019). In order to understand the role of complex microbes on human health, animal models have been developed and used to study the causes and mechanisms underlying various diseases (Kennedy et al., 2018). A recent study using germ-free mice reported that gut microbiota (i.e., all living members forming the microbiome) influence physiological functions and can cause diseases and serious medical conditions, such as artery thrombosis (Hasan et al., 2020). However, no studies to date have investigated the uterine microflora using sterile mouse models.
In an effort to identify most of the microorganisms living in the body including the urogenital tract, the National Institute of Health has initiated a large-scale (300 healthy volunteers) human microbiome project (Peterson, 2009). However, while most earlier research focused on analyzing microorganisms using a culture-dependent approach, current microbiome studies are based on next-generation sequencing techniques such as marker gene amplicon and shotgun sequencing (Verhelst et al., 2004; Franasiak et al., 2016; Knight et al., 2017). Such technological advances have enabled research into the microbiome of the uterus, which was thought to be sterile (Franasiak et al., 2016; Perez-Muñoz et al., 2017). A number of studies have investigated the vaginal microbiota, whereas research on the microbiota of the upper genital tract is scarce; there are only some reports on core taxa in the human uterus (Mitchell et al., 2015; Verstraelen et al., 2016; Tao et al., 2017). Recent research based on culture-independent methods indicated the presence of complex microbial community in the uterus environment, but showed that microbial density is lower than in the intestines (Perez-Muñoz et al., 2017; Baker et al., 2018; Benner et al., 2018; Bai et al., 2021). Furthermore, it has been reported that the microorganisms present in the endometrium act as physiological regulators and can affect the implantation of an embryo (Benner et al., 2018). Studies on animal models are therefore needed to better understand the influence of microbiota on the uterus.
In this study, we aimed to examine the bacterial community of the germ-free mice uterus, which we induced by feeding animals a combination of antibiotics. We compared the bacterial community structures of uteri obtained using 16S rRNA gene amplicon sequencing in control and germ-free mice to identify the characteristics of uterus microbiota under antibiotic selection pressure. This study provides insights into changes in the intrauterine microbiome and their potential effects on embryo implantation capacity that can inspire and guide further research.
MATERIALS AND METHODS
All animal experiments used 6-week-old female ICR mice purchased from Hanil Laboratory Animal Center of Korea. They were housed in the animal facility of Jeonbuk National University, at a temperature of 23±1°C and a humidity of 50±2%, and under a 12 h:12 h light-dark cycle. The mice had free access to a standsard rodent diet and water ad libitum and were adapted to the experimental environment for 3 days prior to antibiotic administration. All experiments were approved by Jeonbuk National University’s Institutional Animal Care and Use Committee (IACUC) (license number: JBNU 2022-050) and in line with our institutional guidelines for the care and use of laboratory animals.
Ten mice were randomly divided into a control and a germ-free group. For the enteral induction of a germ-free environment, one group was given tap water with a combination of antibiotics for 10 days: dissolved colistin sulfate (6,000 U/mL, Duchefa Biochemie, St. Louis, MO, USA), vancomycin hydrochloride (0.5 g/L, Duchefa Biochemie), ampicillin trihydrate (0.1 g/L, Sigma-Aldrich, St. Louis, MO, USA), and neomycin sulfate (1 g/L, Daejung, Siheung, Korea). The antibiotics mix was stored at 4°C, and the drinking water for the germ-free group was changed every day.
Mice were sacrificed through CO2 asphyxiation and uteri were removed under aseptic conditions. Removed uterus samples were snap-frozen using liquid nitrogen and stored at −80°C until DNA was isolated.
Microbial community DNAs were extracted from the 10 uteri (from five controls and five germ-free animals) using the homogenizing mechanical disruptor FastPrep24 and the FastDNA™ Spin Kit for Soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturers’ instructions. Additionally, in order to confirm the antibiotics-induced reduction of the intestinal microflora, total DNA was isolated from colon samples of three mice in each group following the aforementioned protocol. DNA quality was examined by agarose gel electrophoresis and purity analyses using A NanoDrop (Biotek, Winooski, VT, USA). Table 1 lists the details for each sample.
The extracted DNA was sent to Macrogen for 16S rRNA amplicon sequencing. Only samples that passed the double-stranded DNA-specific fluorometry quality control were used. Amplicon libraries targeting the 16S rRNA V3-V4 region were prepared using the universal primers 341F (5’-CCT ACG GGN GGC WGC AG-3’) and 805R (5’-GAC TAC HVG GGT ATC TAA TCC-3’) (Herlemann et al., 2011). The Agilent Technologies 2100 Bioanalyzer (Macrogen, Seoul, Korea) was used to verify the size of the PCR-enriched fragments. The libraries underwent 300-bp paired-end sequencing on a MiSeq platform (Illumina, San Diego, CA, USA) at Macrogen.
Real-time PCR analyses were conducted in duplicate using the TOP real qPCR 2× PreMIX PCR kit (Enzynomics, Daejeon, Korea) and a Rotor-Gene Q thermal cycler (Qiagen, Hilden, Germany) in the following sequence: initial denaturation at 95°C for 15 min, and 40 cycles at 94°C for 10 s, at 60°C for 10 s, and at 72°C for 20 s. Expression levels were normalized to the mammalian gDNA sequence, Crebzf gene. The following primers were used: Crebzf forward, 5’-CTG CCC GTC TTA ATC GGC TC-3’; Crebzf reverse, 5’-CCG TAG GTA GCG ACT CTC CTC-3’; 16S rRNA gene forward (341F); 16S rRNA gene reverse (785R), 5’-GAC TAC HVG GGT ATC TAA TCC-3’.
To assess differences in bacterial community structures in mouse uteri between the control and the germ-free group, the raw 16S rRNA sequence reads were submitted to the EzBioCloud database (http://ezbiocloud.net; CJ Bioscience, Seoul, Korea) (Yoon et al., 2017). We used the 16S rRNA gene-based microbiome taxonomic profiling (16S-based MTP) program embedded in the database for our analyses. Low-quality and chimeric reads were removed and only valid sequences were further analyzed (Table 1). The reads were clustered into operational taxonomic units (OTUs) using 0.03% as a cut-off. The PKSSU4.0 database included in EzBioCloud was used for the taxonomic assignment of all reads. Bacterial compositions were assessed, α-diversity indices were calculated (Chao1, ACE, Shannon, and Simpson), a principal coordinate analysis (PCoA) was applied based on a UniFrac dissimilarity matrix, and statistical comparisons between the groups were conducted using the 16S-based MTP program.
RESULTS
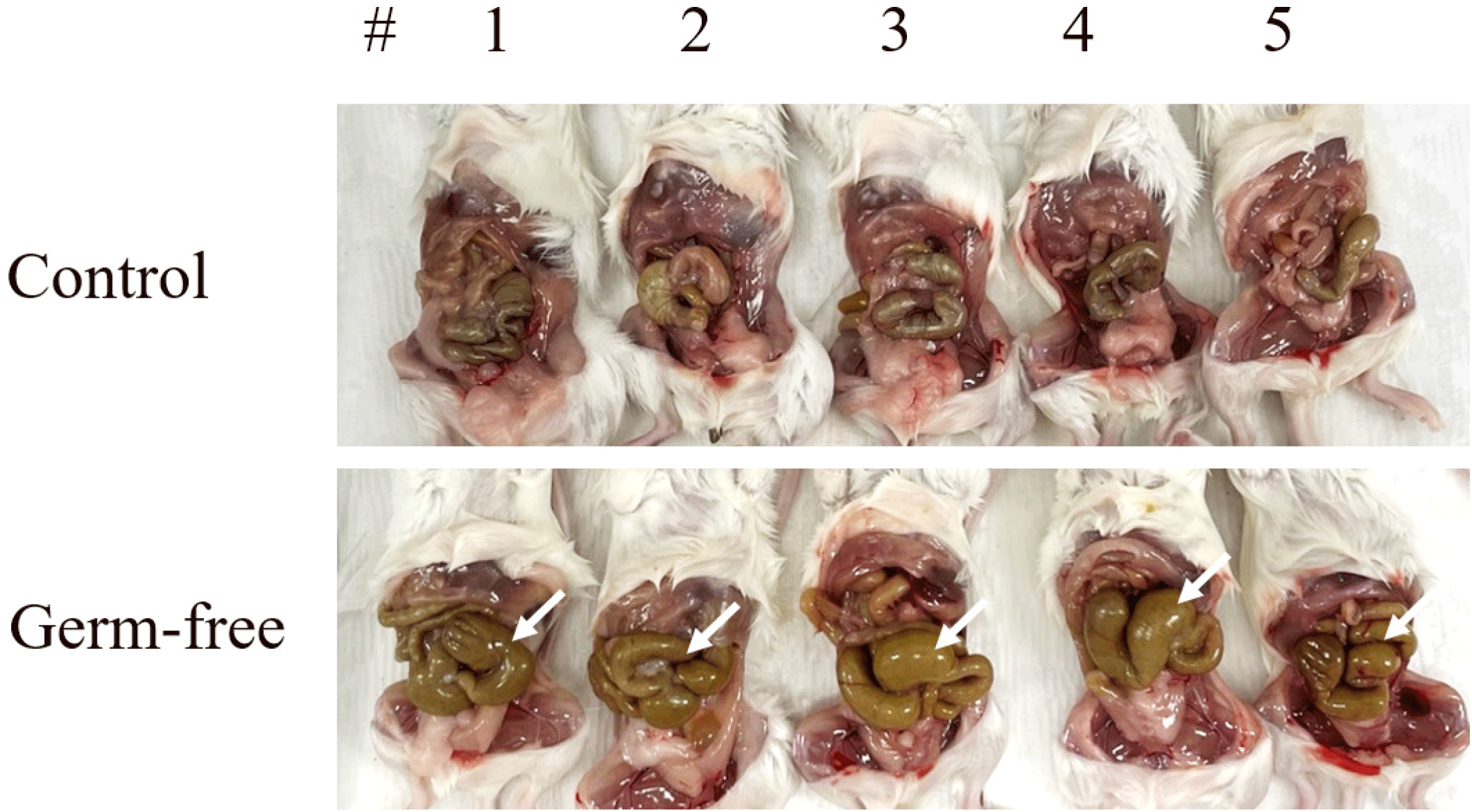
To create mice with sterile intestines, the antibiotics mixture was fed to 6-week-old mice ad libitum for a total of 10 days. We observed intestinal distension, which confirmed the development of a germ-free environment (Fig. 1).
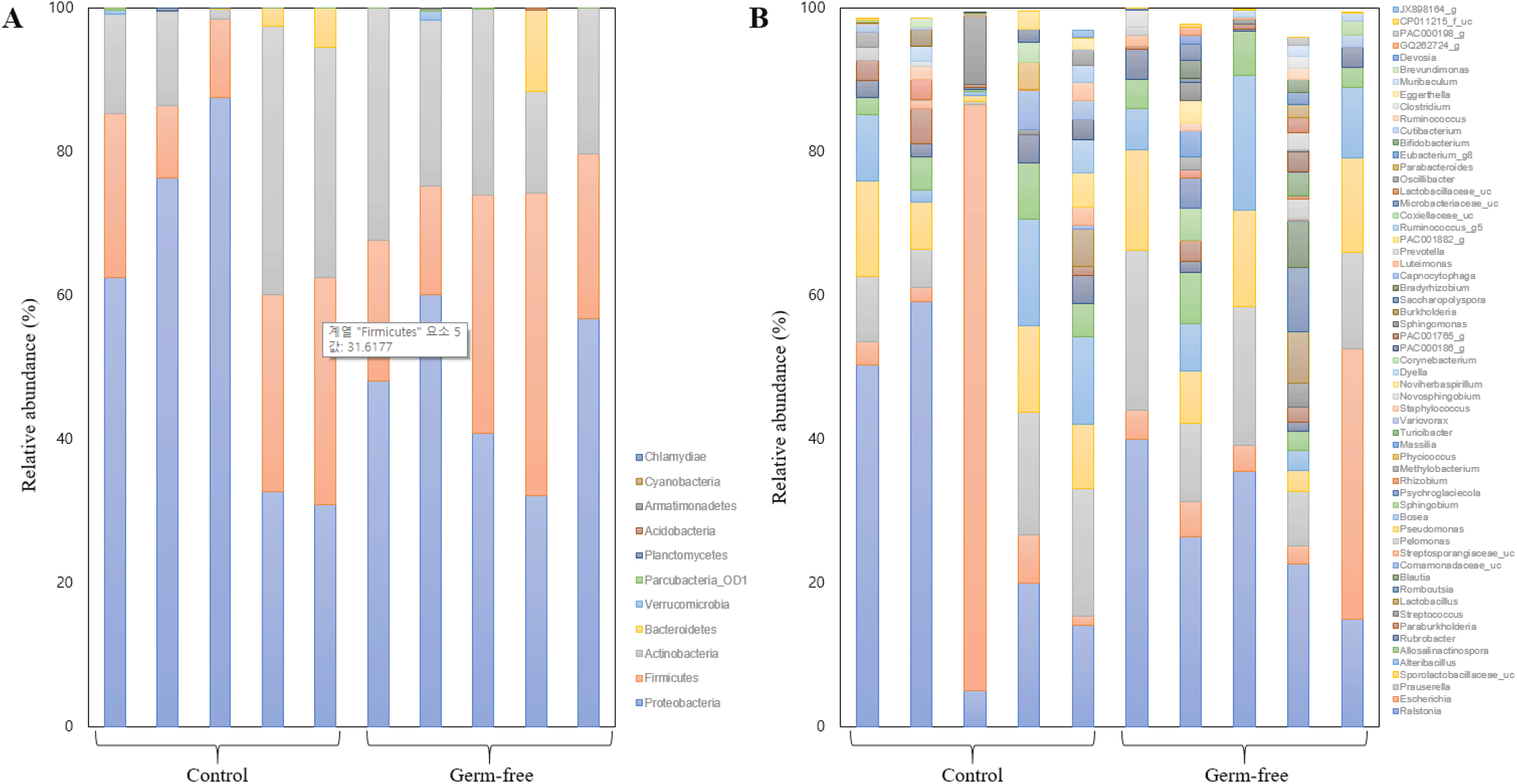
Prior to our community structure analyses, rarefaction curves were generated using the number of OTUs per the number of reads to confirm that the sequencing depth was sufficient to assess the bacterial composition of the mouse uterus (Table 1). The accumulated number of OTUs/reads reached a plateau in each sample (data not shown). Sub-sampling was performed to normalize all samples, based on the lowest read number (10,956). Fig. 2 shows the dominant taxa revealed by our analyses. Both the control and the germ-free group shared three major phyla, i.e., Proteobacteria, Firmicutes, and Actinobacteria, while the bacterial community in the human uterus harbors the phylum Bacteroidetes as another dominant member (Baker et al., 2018). At the genus-level, Ralstonia, Escherichia, and Prauserella were found to be dominant (Fig. 2). The average relative abundances of major taxa in the control group were as follows: Proteobacteria (57.74%), Firmicutes (20.72%), and Actinobacteria (19.55%) at the phylum-level, and Ralstonia (29.64%), Escherichia (18.81%), and Prauserella (9.93%) at the genus-level. In the germ-free group, the dominant phyla were Proteobacteria (47.47%), Firmicutes (26.64%), and Actinobacteria (23.16%), and the major genera Ralstonia (27.87%), Prauserella (14.75%), and Escherichia (10.53%).
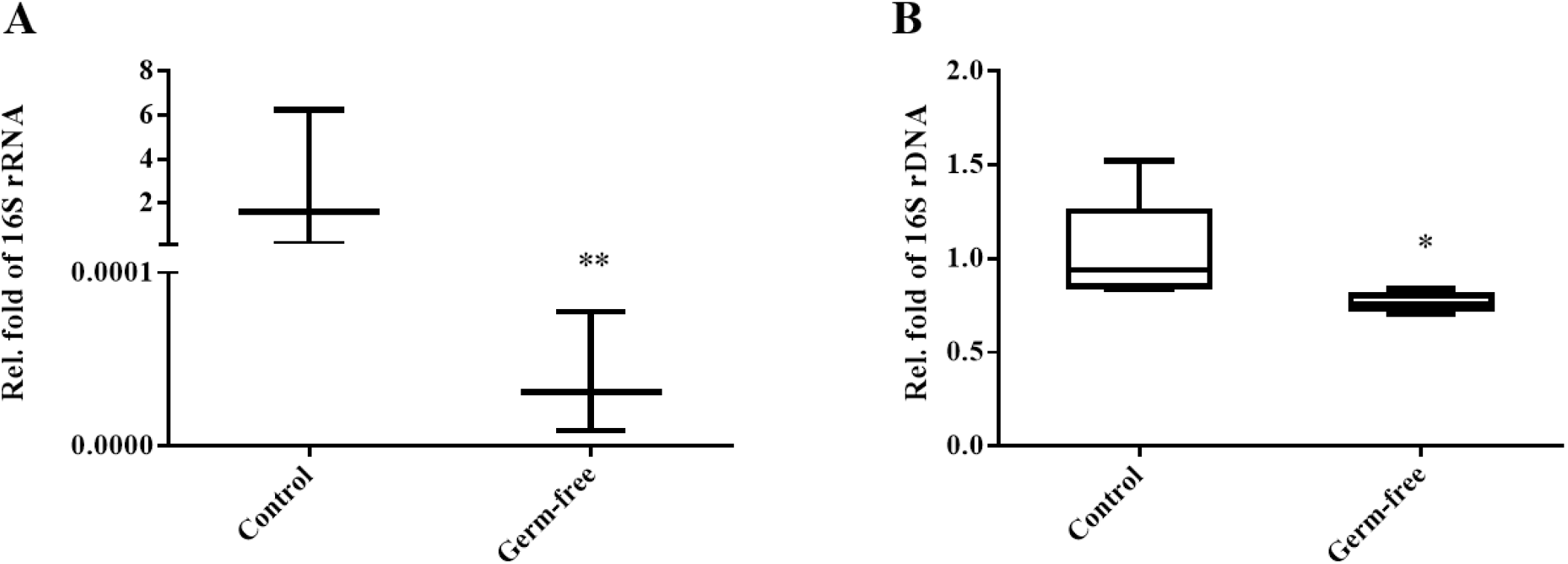
Bacterial population density was analyzed by assessing a bacterial marker gene (16S rRNA gene) in the community DNA extracted from colon and uterus samples using a real-time PCR platform. Microbial population density was normalized using the crebzf gene as a reference. We confirmed that the colons of mice in the antibiotic intake group were clearly germ-free (Fig. 3A). Moreover, overall bacterial density was significantly reduced in the uteri of germ-free animals compared to those obtained from the control group (Fig. 3B). Taken together, these results suggest that antibiotics-induced intestinal sterility can affect not only the intestinal but also the uterine bacterial community.
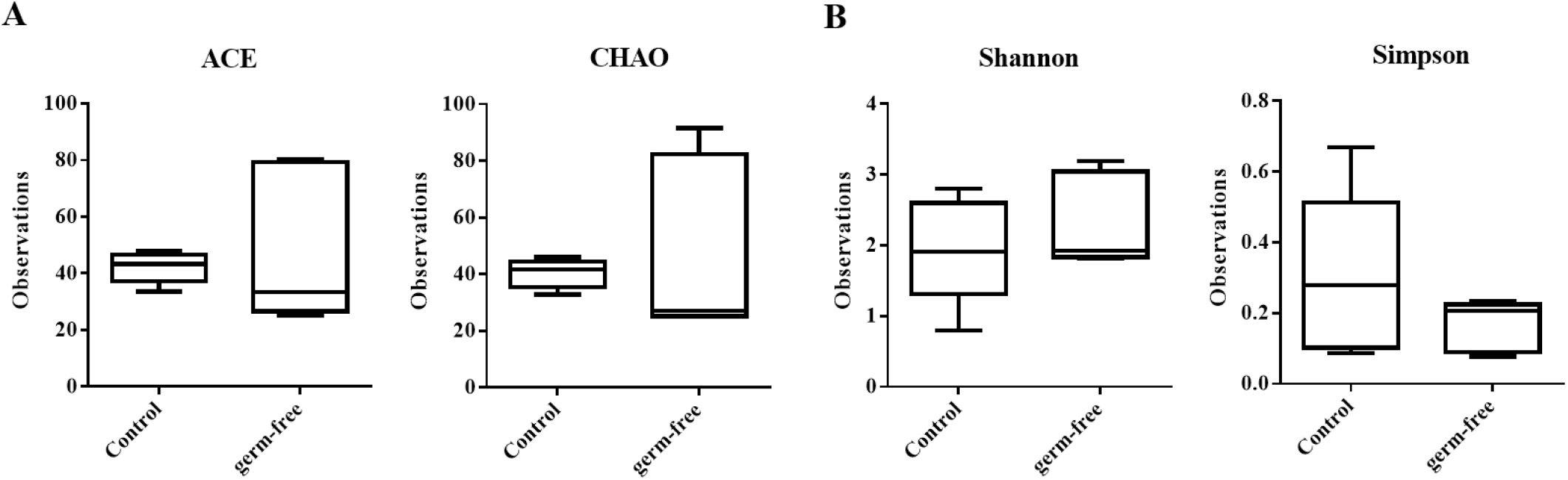
We mined the data to assess whether the reduction in bacterial population density we observed was caused by changes in specific taxa, but identified no populations showing statistically significant differences between the mouse groups. These results are in line with those on α-diversity, which also indicate no difference in species richness and diversity between the groups (Fig. 4). We therefore postulate that the observed diminishing in the bacterial community was caused by an overall reduction in members rather than a proliferation of some and a decline in other bacteria under antibiotic selection pressure.
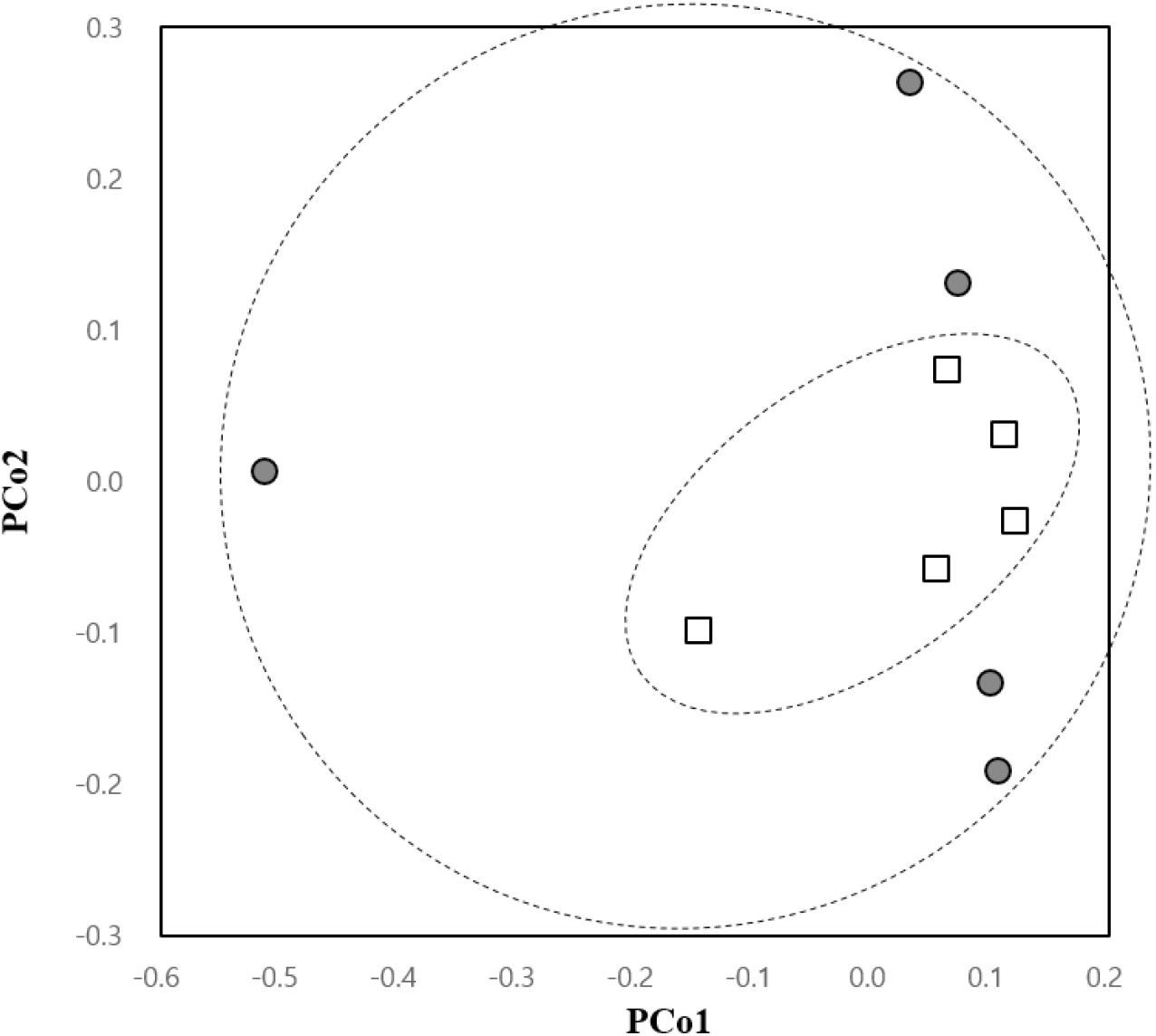
Remarkably, the β-diversity analysis showed a trend in the change of uterine microbial community in the antibiotic-treated group. The PCoA plot indicated more convergent bacterial clustering in the germ-free compared to the control group, although the difference was not statistically significant (Fig. 5). The sporadic pattern observed in the control group seemed to be a result of individual differences within the group, while the converging pattern seen in sterile mice indicated a uniformization of the community structure accompanied by a reduction in population density.
DISSCUSSION
A number of studies have focused on the intestine microbiota of mice models under various selection pressures, including feeding with antibiotic cocktails to create animals with sterile intestines (Theriot et al., 2014; Livanos et al., 2016; Suez et al., 2018; Gobbo et al., 2022; Li et al., 2022). Because the uterine microbial community is deeply related to the gut microbiome, studies on changes in the uterine microenvironment in antibiotic-mediated germ-free mice are needed. In this study, we analyzed the effect of the administration of an antibiotic cocktail on the microbiota of the mouse uterus. The microbiome of the uterus is still not well known, but an effect of microorganism composition on embryo implantation rates has been suggested (Franasiak et al., 2021). Because the dominant taxa of the mouse uterus have not been identified so far, we first determined the general bacterial community structures of mouse uteri at the phylum- and genus-level. We found that the mouse uterus contains the major phyla Proteobacteria, Firmicutes, and Actinobacteria (Fig. 2A), while the human uterus harbors Bacteroidetes as an additional dominant phylum (Baker et al., 2018). At the genus-level, the major taxa in the mouse uterus are Ralstonia, Prauserella, and Escherichia (Fig. 2B). While Ralsotonia and Prauserella do not seem to be core members of the human uterus microbiome (Benner et al., 2018) Ralstonia has been identified as the most abundant genus of the normal microflora in specific mouse models (Bai et al., 2021).
Several studies have shown that antibiotics affect the population density of bacterial communities in animal intestines as well as their diversity indices (Shojaee AliAbadi & Lee, 2000). We also found a dramatic decrease in bacterial density in the mouse intestine following antibiotic treatment (Fig. 3A). The microbiome of the uterus, however, appeared to be less affected than the gut microbiome, which was directly exposed to the antibiotics. Our results suggest that antibiotic treatment cannot radically change the bacterial composition of the mouse uterus and cannot enrich or eliminate specific taxa within the community. However, it should be noted that the administration of an antibiotic mixture resulted in the elimination of variations across individuals in microbiota composition, indicating that the treatment can be used to ensure a uniform background.
Research to date has mainly focused on the gut microbiota of mice. Still, the study of the uterine microbiome of model organisms is essential to address specific challenges in the research of uterus function. This study provides preliminary evidence that should be further explored in future research covering the complicated characteristics and roles of the mouse uterus microbiota.