
INTRODUCTION
Natural killer (NK) cells are cytotoxic lymphocytes that are capable of killing virus-infected cells and cancer cells. Unlike CD8+ T cells, which have cytotoxic activity after adaptive differentiation by the recognition of antigen peptides bound to major histocompatibility complex (MHC)-I molecules, NK cells do not require a differentiation process to eliminate contacted target cells and display rapid cell-mediated cytotoxicity. Therefore, NK cells are regarded as immune cells that function as the first line of defense against cancer cells and viral infections ( Janeway & Medzhitov, 2002). The cytotoxic ability of NK cells to recognize and kill target cells is controlled by the integration of signals that result from various combinatory contacts between cell surface receptors on NK cells and matching ligands on target cells. NKG2D is an activating receptor on NK cells (Bauer et al., 1999), and stimulation of NK cells by NKG2D ligands induces activation of various signals, including the phosphatidylinositol 3-kinase (PI3K) and c-JUN N-terminal kinase ( JNK) MAP kinase pathways (Sutherland et al., 2002; Li et al., 2008). In particular, activation of JNK kinase is required for the movement of the microtubule organizing center (MTOC) and granule polarization in activated NK cell cytotoxicity (Li et al., 2008).
In addition to target recognition by receptor-ligand interactions, the cytotoxicity of NK cells is modulated by other factors, including interleukins (IL-12 and IL-15, for example) (Huntington., 2014; Sim et al., 2014; Ge et al., 2017). NK cells are also activated by phorbol 12-myristate 13-acetate (PMA) and the ionophore ionomycin (ION), which lead to increased NK cell degranulation (Romera-Cárdenas et al., 2016). Many compounds have also been identified as activators of the protein kinase C (PKC) signaling pathway, which is a major lytic signaling pathway in NK cells (Rana & Whalen., 2015). Stimulation of NK cells by exogenous factors generally results in increased killing of target cells, increased levels of degranulation or increased interferon-γ production.
ELK3 is a proto-oncogenic transcription factor of the ETS family and is a therapeutic target in various cancers, including breast cancer and liver cancer (Kong et al., 2016; Lee et al., 2017). XRP44X is a pyrazole chemical that inhibits the transcriptional activity of ELK3. XRP44X inhibits Ras signaling-mediated phosphorylation of ELK3, and the application of XRP44X results in various cellular effects, including G2-M cell cycle arrest and suppression of angiogenesis (Wasylyk et al., 2008). In addition, XRP44X acts as a microtubule depolymerization agent by activating the JNK pathway (Chen et al., 2012). Administration of XRP44X to preclinical mouse tumor model resulted in the inhibition of tumor growth and metastasis (Semenchenko et al., 2016).
Since the ETS family transcription factor ETS1 regulates the expression of signaling molecules that are essential for NK cell activation and Ets1−/− NK cells are in a chronically activated state (Ramirez et al., 2012), we examined whether the application of XRP44X, an inhibitor of the ETS family gene ELK3, modulated the cytotoxic activity of NK cells.
We found that XRP44X stimulated NK cells to enhance cytotoxic activity against breast cancer cells. In the presence of XRP44X, NK cells did not show notable apoptosis or impaired cell cycle progression. We demonstrated that XRP44X enhanced the cytotoxicity of NK cells through stimulation of interferon gamma expression and activation of the JNK signaling pathway. Our data suggest that XRP44X has the potential to be applied in developing NK cells as an immune-oncogenic therapeutic.
MATERIALS AND METHODS
NK-92MI and MDA-MB231 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The human NK cell line NK-92MI was cultured in Minimum Essential Medium alpha (MEM-α, Invitrogen, Carlsbad, CA, USA) supplemented with 2 mM L-glutamine (Invitrogen), 1.5 g/L sodium bicarbonate (Sigma-Aldrich, St. Louis, MO, USA), 0.2 mM myo-inositol (Sigma-Aldrich), 0.1 mM 2-mercaptoethanol (Invitrogen), 0.02 mM folic acid (Sigma-Aldrich), 12.5% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). XRP44X was purchased from Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO, Invitrogen).
To detect apoptosis, NK-92MI cells (5×105) were cultured in a 12-well cell culture plate and treated with XRP44X. After an additional 48 hr of culture, the cells were harvested and resuspended in an appropriate volume of cold phosphate-buffered saline (PBS, Invitrogen) for analysis. Treated cells were stained by using an annexin V-FITC/PI apoptosis kit (Invitrogen) to monitor apoptotic cells. The collected cells were obtained and analyzed using a CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA).
NK-92MI cells were treated with XRP44X for 48 hr, harvested, washed, resuspended in 70% ethanol and stored at 4°C overnight. Then, the cells were suspended in cold PBS. Subsequently, the cells were incubated with 0.1 mg/mL RNase I and 50 mg/mL PI at 37°C for 30 min. The cell cycle was then detected with a CytoFLEX flow cytometer (Beckman Coulter).
MDA-MB231 cells (1×105) were prestained with CellTrace CFSE (Invitrogen) and cocultured with NK-92MI cells at a 10:1 effector:target (E:T) ratio. After 4 hr of coincubation, all cells were harvested and stained with 7-AAD (Invitrogen) for live/dead discrimination. The samples were analyzed on a CytoFLEX flow cytometer (Beckman Coulter). Triplicate experiments were performed.
The cytotoxicity of NK cells was also determined using a CytoTox-Glo™ cytotoxicity assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. After 4 hr of coincubation of NK-92MI and MDA-MB231 cells, 50 μL of CytoTox-Glo™ cytotoxicity assay reagent was added to the reactions, and the luminescent signal that reflects the cytotoxicity was measured using a SpectraMax L microplate reader (Molecular Devices, Sunnyvale, CA, USA). Cytotoxicity was calculated by dividing the luminescent dead-cell signal by the total cell luminescence value.
Total RNA from NK-92MI cells was extracted using TRIzol reagent (Invitrogen). One microgram of total RNA was treated with RNase-free DNase I (Invitrogen), and cDNA was prepared using the SuperScript II First-Strand Synthesis System (Invitrogen). Quantitative RT-PCR was performed with iQ SYBR Green PCR Mastermix (Bio-Rad, Hercules, CA, USA).
For protein analysis, cells were washed twice with cold PBS (Invitrogen) and lysed in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA). Total cell extracts were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad), and blotted with antibodies against GAPDH, phospho-JNK, JNK, phospho-NF-κB, NF-B, phospho-p38, p38, phospho-AKT, AKT, phospho-ERK1/2, ERK1/2 (Cell Signaling Technology), ELK3 (Novus, Centennial, CO, USA), and phospho-ELK3 (GeneTex, Irvine, CA, USA). Immunoreactivity was detected with enhanced chemiluminescence (Thermo Fisher Scientific, Rochester, NY, USA).
To determine the level of IFN-γ in XRP44X-treated NK-92MI cell culture media, ELISA was performed. All reagents were obtained from BD (Franklin Lakes, NJ, USA). Immunoplates were coated overnight at 4°C with coating antibodies and buffer. Plates and reagents were brought to room temperature, and the plate was washed three times with washing buffer. The plates were blocked for 1hr at room temperature with 100μL blocking buffer. Then, the samples and standards were added to each well for 2 hr at room temperature. After washing, 100μL of detection antibody solution was added to each well for 1hr at room temperature. For color development, 50μL TMB was added to each well. The plates were incubated for 30 min in the dark before the reaction was stopped by adding 50μL of stop solution to each well. The absorbance was mEeasured at 450nm using a microplate reader.
Samples were analyzed with Student’s t-test or ANOVA with Duncan’s multiple range procedure for multiple comparisons. All statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad Software, USA) or the SigmaPlot 11.2 program (Systat Software, USA). The error bars represent the standard errors from three independent experiments, which were each performed using triplicate samples. p-values less than 0.05 were considered statistically significant.
RESULTS
To determine the concentration of XRP44X that was not cytotoxic, we first examined the effect of various concentrations of XRP44X on the cell cycle of the human NK cell line NK-92MI. As shown in Fig. 1A, 0.8 μM XRP44X induced a slight increase in the G2/M population, which is consistent with the activity of XRP44X in MCF-7 breast cancer cells (Chen et al., 2012).
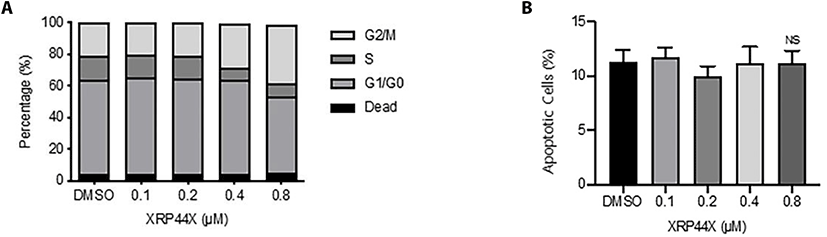
Although 0.8 μM XRP44X affected the cell morphology of MDA-MB231 cells, NK cell morphology was not affected by the same concentration of XRP44X (data not shown). To further confirm whether cell viability was affected, we performed annexin V/PI staining analysis. Consistent with the cell cycle analysis, 0.8 μM XRP44X did not increase the dead cell population of NK-92MI cells (Fig. 1B). Based on these results, we concluded that XRP44X does not have toxic effects on NK-92MI cells up to a concentration of 0.8 μM, and we further analyzed the effect of XRP44X on the cytotoxic activity of NK cells against cancer cells at a concentration of 0.8 μM.
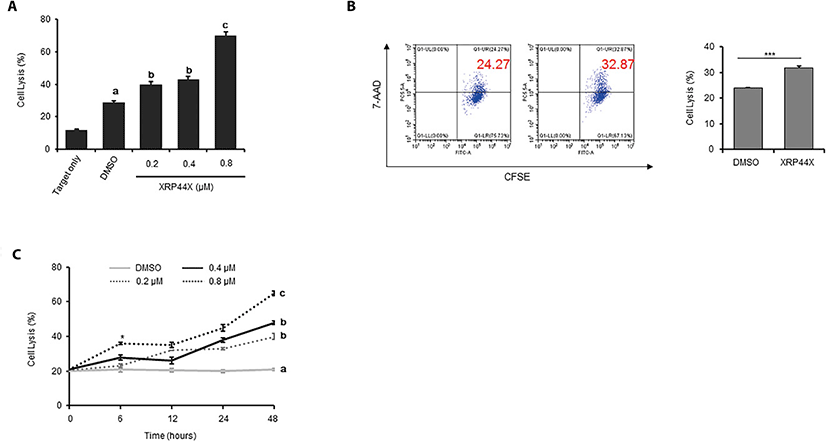
To analyze whether XRP44X increases the cytotoxic activity of NK-92MI cells toward the triple negative breast cancer cell line MDA-MB231, we pretreated NK-92MI cells with XRP44X for 48 hrs and then cocultured the cells with MDA-MB231 cells for 4 hrs. When the cytotoxicity of NK-92MI was quantified using a CytoTox Glo™ cytotoxicity assay that selectively measures dead-cell protease activity, it was clearly revealed that the cytotoxic activity of NK-92MI cells against MDA-MB231 cells gradually increased in a XRP44X concentration-dependent manner (Fig. 2A). The effect of XRP44X on the cytotoxicity of NK-92MI was further supported by flow cytometry analysis with CFSE/7-AAD staining. Similar to the dead cell protease analysis results, the target cell lysis ability of NK-92MI was increased by treatment with 0.8 μM XRP44X (Fig. 2B). By analyzing the cytotoxicity of NK cells that were pretreated with XRP44X for various times, we identified that the effect of XRP44X on NK-92MI cells gradually increased depending on the pretreatment time, and we also revealed that 0.8 μM XRP44X increased NK cell activity with as little as 6 hr of pretreatment (Fig. 2C).
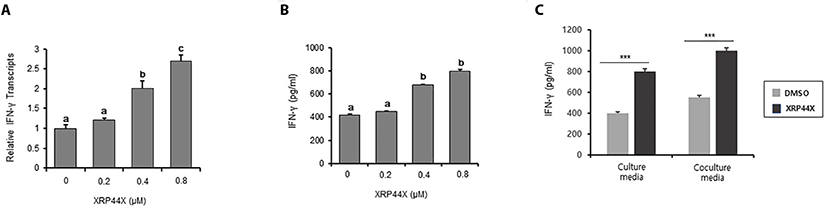
Since NK cells exhibit a cytotoxic immune response by producing IFN-γ, we next examined whether XRP44X stimulates NK cells to produce increased amounts of IFN-γ. As expected, IFN-γ transcripts were significantly enhanced by treatment with 0.4 μM and 0.8 μM XRP44X (Fig. 3A). Consistently, secreted IFN-γ in NK cell culture media was increased in a XRP44X concentration-dependent manner (Fig. 3B). The amount of secreted IFN-γ was further increased when XRP44X-treated NK-92MI cells were cocultured with MDA-MB231 cells (Fig. 3C). These results suggest that the mechanism by which XRP44X increases the cytotoxic activity of NK-92MI cells toward MDA-MB231 cells is by stimulating IFN-γ expression and secretion.
The mitogen-activated protein kinase (MAPK) signaling pathway is reported to be activated during NKG2D-mediated NK cell cytotoxicity (Li et al., 2008). To examine whether the MAPK signaling pathway is activated by XRP44X treatment, we examined the phosphorylation status of c-JUN N-terminal kinase ( JNK), p38 and extracellular signal-regulated protein kinase (ERK) following XRP44X treatment. As shown in Fig. 4A, JNK was significantly phosphorylated by 0.8 μM XRP44X, whereas p38 and ERK1/2 were not phosphorylated under the same conditions. To determine whether activation of JNK by XRP44X is associated with NK activation, we examined the effect of chemical inhibition of JNK phosphorylation on XRP44X-mediated activation of NK cells. As expected, the degree of NK activation by XRP44X was significantly decreased when NK cells were cotreated with the JNK inhibitor SP600125 to inhibit XRP44X-mediated phosphorylation of JNK (Fig. 4B-C).
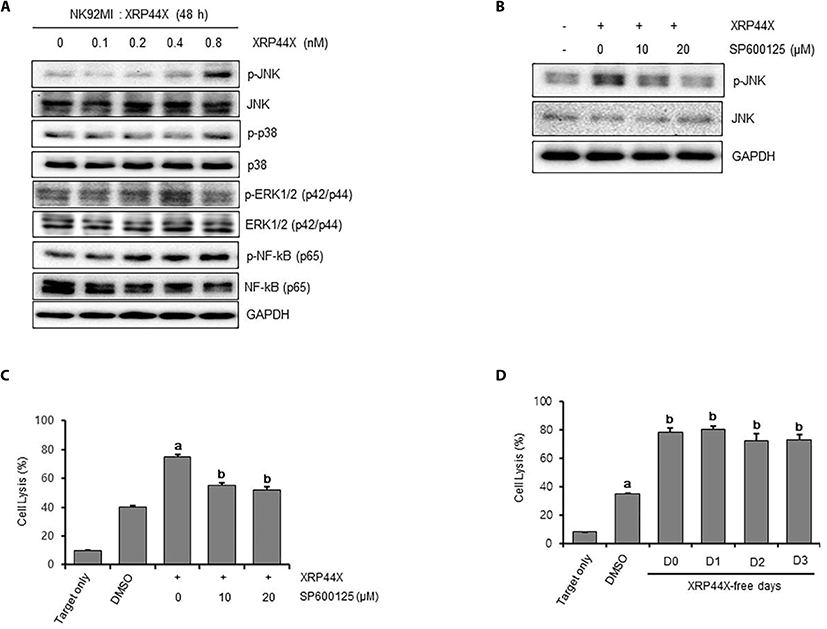
Finally, we examined whether XRP44X-mediated activation of NK cells sustains their enhanced activity in the absence of XRP44X. For this purpose, we first cultured NK-92MI cells for 48 hr in the presence of XRP44X and then transferred the cells to fresh media without XRP44X. As shown in Fig. 4D, NK cells that were activated by XRP44X maintained their enhanced cytotoxic activity against MDA-MB231 cells up to three days after the removal of XRP44X. Taken together, we concluded that XRP44X activates naïve NK cells in the absence of target cells and that XRP44X-mediated activation of JNK signaling is one of the major mechanisms that explains the enhanced activity of NK cells.
DISCUSSION
The ETS family transcription factor ETS1 regulates the expression of signaling molecules that are essential for NK cell activation, and Ets1-/- NK cells are in a chronically activated state (Ramirez et al., 2012). Based on this report, we examined whether ELK3, another ETS family transcription factor, is associated with the cytotoxic activity of NK cells. The pyrazole chemical XRP44X was identified as a candidate anticancer drug that inhibits ELK3 activity in various cells, including cancer cells (Wasylyk et al., 2008; Chen et al., 2012; Semenchenko et al., 2016), and we applied XRP44X to NK cells.
Pyrazole is a type of chemical that has a 5-membered ring structure with 2 nitrogen atoms and aromatic heterocycles. Its derivatives have been applied for a wide range of purposes, including pesticidal, antibacterial, antihepatotoxicity, anti-inflammatory, and analgesic activities. We confirmed in this study that the pyrazole derivative compound XRP44X increases the cell-mediated cytotoxic ability of NK cells. XRP44X is an inhibitor of ELK3, the RAS sub-signal molecule (Semenchenko et al., 2016), and a microtubule depolymerizing reagent (Wasylyk et al., 2008). When we treated the triple negative breast cancer cell line MDA-MB231 with XRP44X, we observed that the cells were condensed and could not maintain their structure (data not shown). This is thought to be the result of XRP44X affecting the cell structure of MDA-MB231 cells.
A recent study of pyrazole compounds on NK cells showed that 4-methylpyrazole (4-MP) activates NK cells (Yi et al., 2015) (1). This finding reveals that 4-MP stimulates NK cells to express IFN-γ, and in our results, a more complex pyrazole, XRP44X, also promoted the expression of IFN-γ at the transcript level. From these studies, we hypothesize that there is a correlation between pyrazoles and IFN-γ expression in NK cells. Usually, IFN-γ is produced when NK cells recognize target cells (Rajagopalan et al., 2001). Our study revealed that XRP44X treatment induces increases of IFN-γ production at resting state of NK cells without target cell contact.
Since RNA level of IFN-γ is increased by XRP44X, it is estimated that transcriptional activation of cytotoxic molecules such as IFN-γ, possibly through the associated signaling pathway, might be one of the main mechanism of XRP44X to enhance anticancer activity of NK cells.
The JNK (c-Jun N-terminal kinase) pathway plays an important role in NK cell activation. There have been reports that the JNK pathway is required for granule depolarization (Li et al., 2008) or is involved in the constitutive expression of CCL5 (Kumar et al., 2009). XRP44X affects various signaling pathways in NK cells, but it is particularly characterized by changes in the phosphorylation of JNK, which is thought to be a factor in increasing NK cell-mediated cytotoxicity. We showed an interesting result regarding the time course activation of JNK and NF-κB. The phosphorylation of JNK dramatically increased with XRP44X treatment and decreased rapidly after XRP44X was removed. On the other hand, phosphorylation of NF-κB increased slowly until the next day and then decreased slowly. This kind of crosstalk between signaling pathways may be an important result for understanding the activation mechanism of NK cells. Since XRP44X was originally identified as an anticancer agent that depolymerized microtubules of cancer cells, our finding that XRP44X enhances the cytotoxic activity of NK-92MI cells presents a new paradigm for the combination of chemotherapy and immunotherapy in tumor treatment.