
INTRODUCTION
Obesity is characterized by excessive body weight and abnormal body fat accumulation and is becoming a serious global health problem (Kopelman, 2000; Chang et al., 2017). It is associated with a chronic inflammatory state and other metabolic disorders such as hyperlipidemia, type 2 diabetes, cardiovascular diseases and cancer (Keller & Lemberg, 2003; Pi-Sunyer, 2009; Han & Lean, 2016). It is associated with an imbalance between energy intake and expenditure and the subsequent excess accumulation and adipogenesis of adipose tissue (Jung & Choi, 2014). Adipogenesis is the process by which an undifferentiated pre-adipocyte is converted into a fully differentiated adipocyte and plays a key role in fat mass growth and the regulation of adipogenesis (Kim et al., 2012; Park et al., 2012; Yang & Kim 2015).
Sirtuins, a family of NAD+-dependent deacetylases, have recently emerged as novel therapeutic targets for metabolic diseases. Sirtuin (silent mating type information regulation 2 homolog) 1 (SIRT1) is thought to influence diverse biological processes via the direct deacetylation of peroxisome proliferator-activated receptor γ (PPARγ), coactivator-1α, forkhead box O1, and p53 and via the indirect activation of AMP-activated protein kinase (AMPK) signaling, by stimulating fat utilization to prevent diet-induced obesity and its associated disorders (Ye et al., 2017). Recently, natural compounds that may be beneficial for reducing the pathogenesis of obesity and metabolic syndrome have been investigated (Graf et al., 2010; Hasani-Ranjbar et al., 2013).
Bitter melon (Momordica charantia L., Cucurbitaceae) grows in many countries of Asia. Its fruit, fruit juice and dried powder preparations have been widely used as traditional medicationsto lowerthe blood glucose levels and obesity andto inhibit the proliferation of various cancer cells by inducing apoptosis (Ray et al., 2010; Fuanchan et al., 2011; Ru et al., 2011). Recently, the ethanol extract of bitter melon (BME) was shown to control pancreatic beta cells in streptozotocin-induced type 2 diabetic rats (Hafizur et al., 2011). We also published a paper that BME had anti-diabetic effect on the high-fat diet (HFD)-fed mice (Yoon et al., 2017). However, how BME interacts with specific targets in the various pathways involved in obesity and metabolic disorders is unknown.
Therefore, in this study, we investigated the potential beneficial effects of BME by using the pre-adipocyte 3T3-L1 cell system and an animal model.
MATERIALS AND METHODS
Preparation of BME was described in a previous study (Yoon et al., 2017). Briefly, BME was manufactured and verified by Daeho Corporation Co., Ltd. (Hwaseong, Korea). All the BME used in present study was grown in Hamyang (Cheonryung Foods Co., Ltd., Hamyang, Korea). The dried unripe fruit was shattered and extracted by heating twice at 70℃ (4 h and 2 h) with 70% ethanol. The extract was then filtered and concentrated (Busung Tech, Ansung, Korea) to 15–20 degrees Brix at 65℃. The concentrated extract was spray dried (Mehyun Engineering, Anyang, Korea) at an inlet temperature of 175±10℃ and an outlet temperature of 80±5℃, and then dextrin (49%) and lecithin (1%) were added. The extraction yield was approximately 34% (w/w).
3T3-L1 pre-adipocyte cells, purchased from ATCC (ATCC CL-173, Manassas, VA, USA), were maintained as pre-adipocytes in 90% Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS (FCS; HyClone, Logan, UT) and 0.1% Pen-Strep (referred to in the text as growth medium). The 3T3-L1 pre-adipocytes were always passaged before the cultures reached 70% confluence. To induce the differentiation of the 3T3-L1 pre-adipocytes to adipocytes, we employed procedures recommended by the ATCC. When the 3T3-L1 pre-adipocytes had reached 100% confluence, the medium was changed to 90% DMEM supplemented with 10% FBS, 1.0 μM dexamethasone, 0.5 mM methylisobutylxanthine and 1.0 μg/mL bovine insulin (referred to as differentiation medium). After 48 h of culture in this medium, the medium was again changed to 90% DMEM supplemented with 10% FBS and 1.0 μg/mL bovine insulin (referred to as maintenance medium). Adipocyte differentiation was determined by oil red O staining. Both pre-adipocytes and adipocytes were cultured in 6-well plates at 37℃ in a humidified atmosphere consisting of 95% air and 5% CO2.
The cells were seeded into 96-well plates at a density of at 1.0×104 cells/well and allowed to attach for 24 h. The cells were treated with vehicle or with the indicated concentrations of BME diluted in complete media for 24, 48, and 72 h. Cell proliferation was determined using MTS assay (Promega, 3580, Madison, WI, USA). The proliferative activities were expressed as the relative percentage at the initial time.
3T3-L1 pre-adipocytes were cultured and fully differentiated with and without BME during 6 days. The cells were washed twice with PBS and fixed with 10% formalin for 30 min. After fixation, they were washed with 60% isopropanol for 5 min and then stained with oil-red O working solution (1.5 mg/mL oil-red O/60% isopropanol) for 15 min at RT. Then, they were washed with distilled water and photographed under a light microscope. Next, the TG levels were measured in cell lysates using Triglyceride Colorimetric Assay Kit (Cayman, Ann Arbor, MI, USA). Cells were washed twice with PBS then scraped and centrifuged at 100×g for 5 min. Cell pellets were lysed in standard diluent buffer and centrifuged at 10,000×g for 10 min. The supernatant was collected and measured the TG.
Cells and tissues were lysed in RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA). Total protein concentrations were determined using the BCA assay (PIERCE, Rockford, IL, USA). An equal volume of 4X SDS sample buffer was added, and the samples were boiled for 5 min. Equivalent amounts of total protein (10–30 μg) were separated by SDS-PAGE on 8%–12% polyacrylamide gel and then transferred to a nitrocellulose membrane. The membrane was blocked with 5% skim milk or 3% BSA in 0.1% Tween-20/Tris buffered saline (TBS-T). The membrane was incubated with SIRT1 (1:1,000, #2493, Cell Signaling Technology, Beverly, MA, USA), PPARγ (1:1,000, #2443, Cell Signaling Technology), sterol regulatory element binding protein 1 (SREBP1) (1:1,000, sc-8984, Santa Cruz Biotechnology, Dallas, TX, USA), AMPK (1:1,000, #2532, Cell Signaling Technology), p-AMPK (1:1,000, #2535, Cell Signaling Technology), and β-actin (1:10,000, A5441, Sigma, St. Louis, MO, USA) primary antibodies overnight at 4℃ and secondary antibodies were incubated for 1 h at RT. Labeled proteins were detected by ECL (Thermo Fisher Scientific) using LAS 4000 (Fujifilm, Tokyo, Japan).
Basically, animal model was same as a previous study (Yoon et al., 2017). Five-week-old male C57BL/6J mice were purchased from Central Lab Animal Inc. (Seoul, Korea). The C57BL/6J mice were randomly divided into four groups (n=8–12). The first group was fed a normal diet (ND), the second was fed a HFD, and the other two groups were fed a HFD supplemented with 250 mg BME/kg BW (BME 250) and 500 mg BME/kg BW (BME 500), respectively. The ND group was fed a chow diet (10% kcal from fat, New Brunswick, NJ, USA, Research Diets; D12450B) or a HFD (45% kcal from fat, New Brunswick, NJ, USA, Research Diets; D12451) for 12 weeks and then fed HFD supplemented with BME 250 (250 mg/kg body weight/day) or 500 (500 mg/kg body weight/day) for 12 weeks. The animals were housed in a temperature-controlled environment with a 12 h light/dark cycle throughout the 12 week experimental period. Food intake and body weight were measured weekly. At the end of the experimental period, animals were anesthetized with zoletil (5 mg/kg, Virbac Laboratories, Carros, France) and perfused with 4% paraformaldehyde solution in 0.1 M phosphate-buffered saline (NBP) through the left ventricle. The white adipose tissues (WATs; epididymal fat, mesentery fat, perierenal fat) were removed and weighed. Portions of WATs were immersed in 4% NBP for histological inspection and other portions were frozen immediately in liquid nitrogen and stored at −80℃ for protein samples. All animal procedures adhered to the Animal Care and Use Guidelines of Gyeongsang National University. The experimental protocol was approved by the Institutional Animal Care and Use Committee of Gyeongsang National University (Jinju, Korea, GNU-140818-M0034).
Fixed WATs in 4% NBP were dehydrated though a graded series of ethanol (70%, 80%, 90%, 95%, and 100% absolute ethanol) for 2 h per concentration, cleared in two changes of xylene for 2 h per change and impregnated using two changes of molten paraffin wax for 2 h per change in a Leica tissue processor (TP 1020, Leica, Wetzlar, Germany). The embedded tissue was sectioned at a thickness of 5 µm with a microtome (RM2235, Leica) and subjected to H&E staining. All stained tissue sections were imaged using a virtual slide microscope (BX61, Olympus, Tokyo, Japan) at ×20 magnification with Olyvia software (Olympus).
The data are expressed as the mean and the standard error of the mean (means±SEM). The differences among the groups were analyzed by one-way analyses of variance (ANOVA), followed by Tukey's HSD test in the GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA, USA).
RESULTS
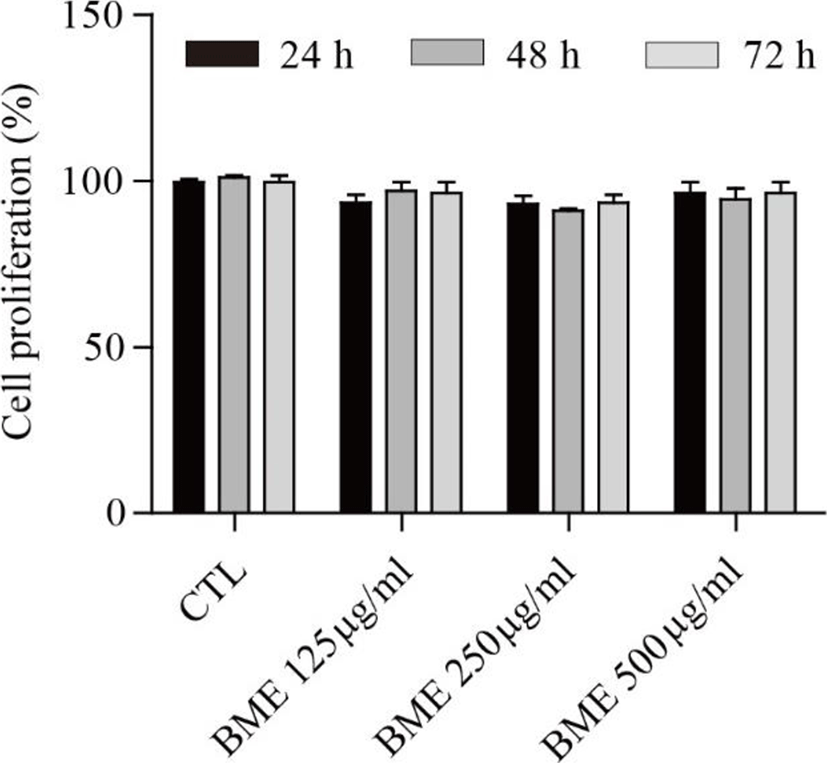
The proliferation assay was performed to assess the effect of the BME on 3T3-L1 cell proliferation. BME showed no significant cytotoxicity at 125, 250, or 500 μg/ml after 24, 48, and 72 h treatments (Fig. 1).
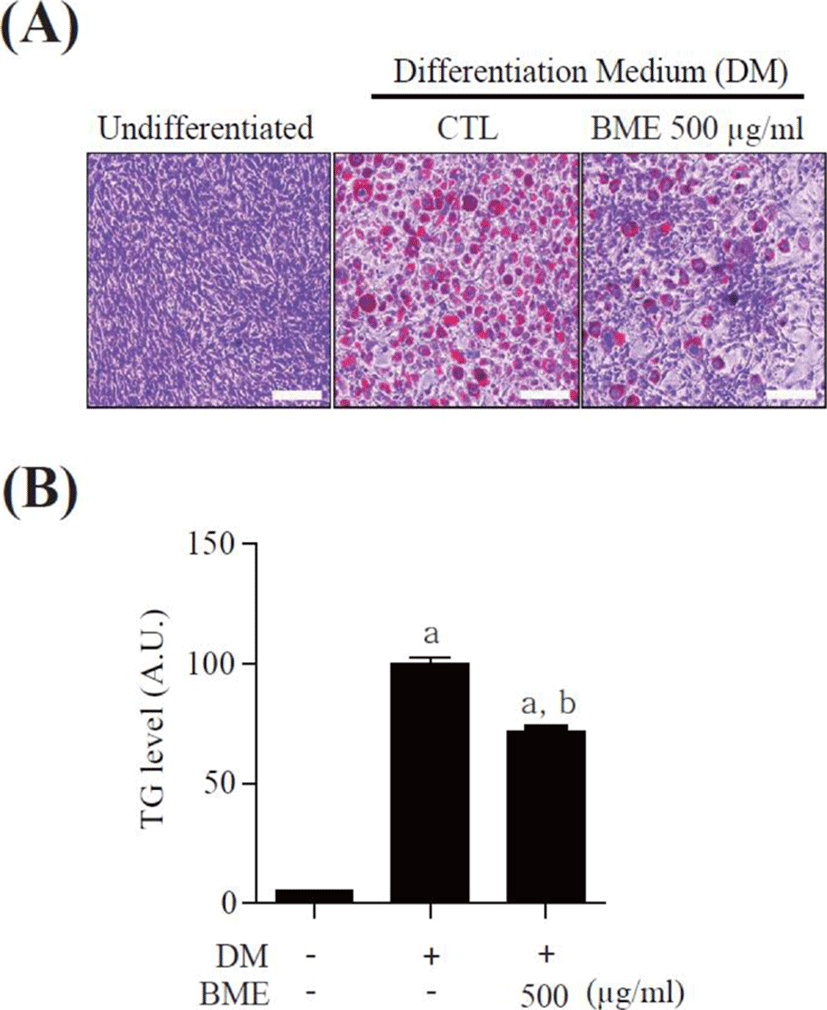
To study the effects of BME on adipocyte differentiation, 3T3-L1 pre-adipocytes were differentiated into mature adipocytes for 6 days in the presence of 500 μg/ml BME and the accumulated intracellular lipids were detected by oil-red O staining. BME 500 μg/ml significantly suppressed lipid drop formation compared with untreated differentiated 3T3-L1 cells (Fig. 2A). The intracellular TG was also reduced in the BME-treated cells (Fig. 2B). The data suggest that BME inhibits 3T3-L1 cells differentiation into adipocyte.
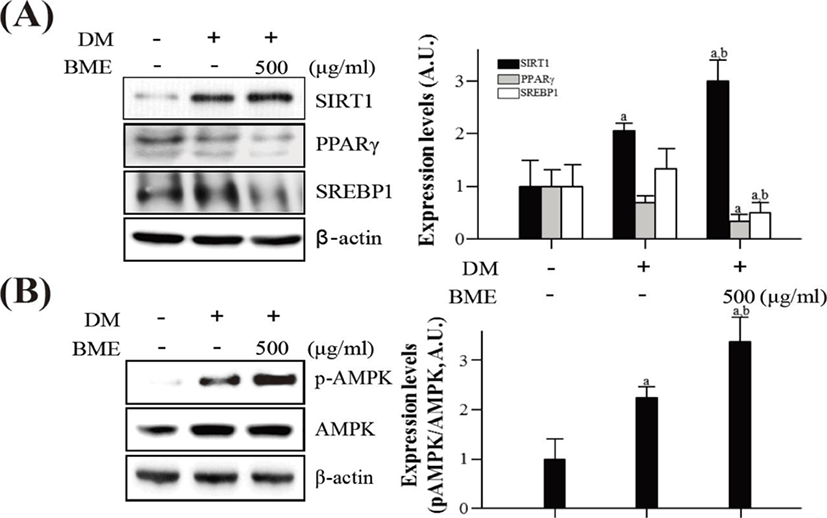
To elucidate the effects of BME on the activation of SIRT1 and AMPK in differentiated 3T3-L1 cells, western blotting was used. BME increased SIRT1 expression and decreased PPARγ and SREBP1 expression (Fig. 3A). Similarly, the BME increased p-AMPK without affecting the protein level of AMPK (Fig. 3B).
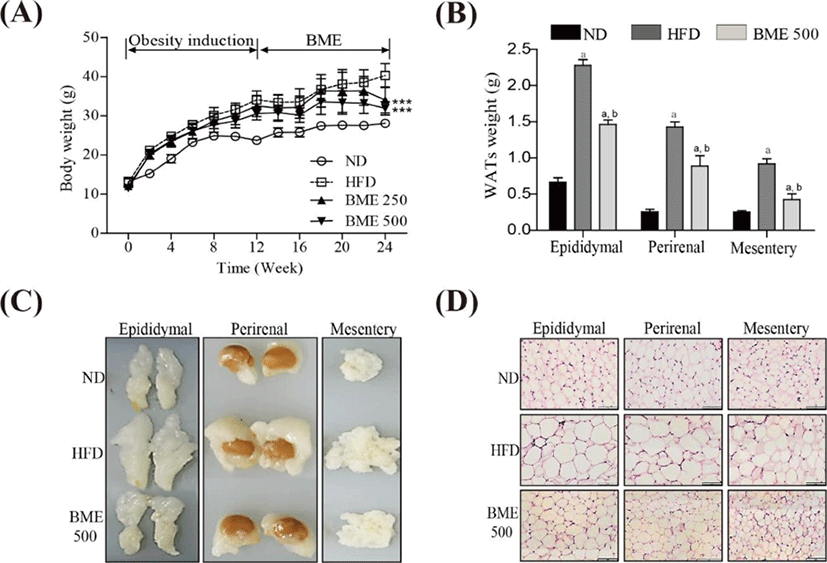
The body weight gain in the HFD group was significantly greater than that in the ND group. Notably, the body weight gain in both the BME 250 and 500 groups were significantly lower than those in the HFD group (Fig. 4A). No significant difference in food intake was observed among the ND, HFD, BME 250 and 500 groups (data not shown), indicating that the reduction in body weight gain in the BME-supplemented HFD groups was not due to reduced caloric intake. To determine whether the reduced body weight gain was related to decreased fat accumulation, WATs were dissected and weighed. The weights of WATs were greater in the HFD group compared to the ND group, but the HFD supplemented with BME 500 group showed reduced increases in WAT weight and size (Fig. 4B, C), indicating that BME ameliorated obesity in a HFD supplemented with BME due to reduced adiposity in WAT. A histological analysis of the WATs revealed smaller adipocytes in the HFD supplemented with BME 500 group than in the HFD group (Fig. 4D), further demonstrating that the reduced body weight gain was due to decreased fat accumulation in the adipocytes.
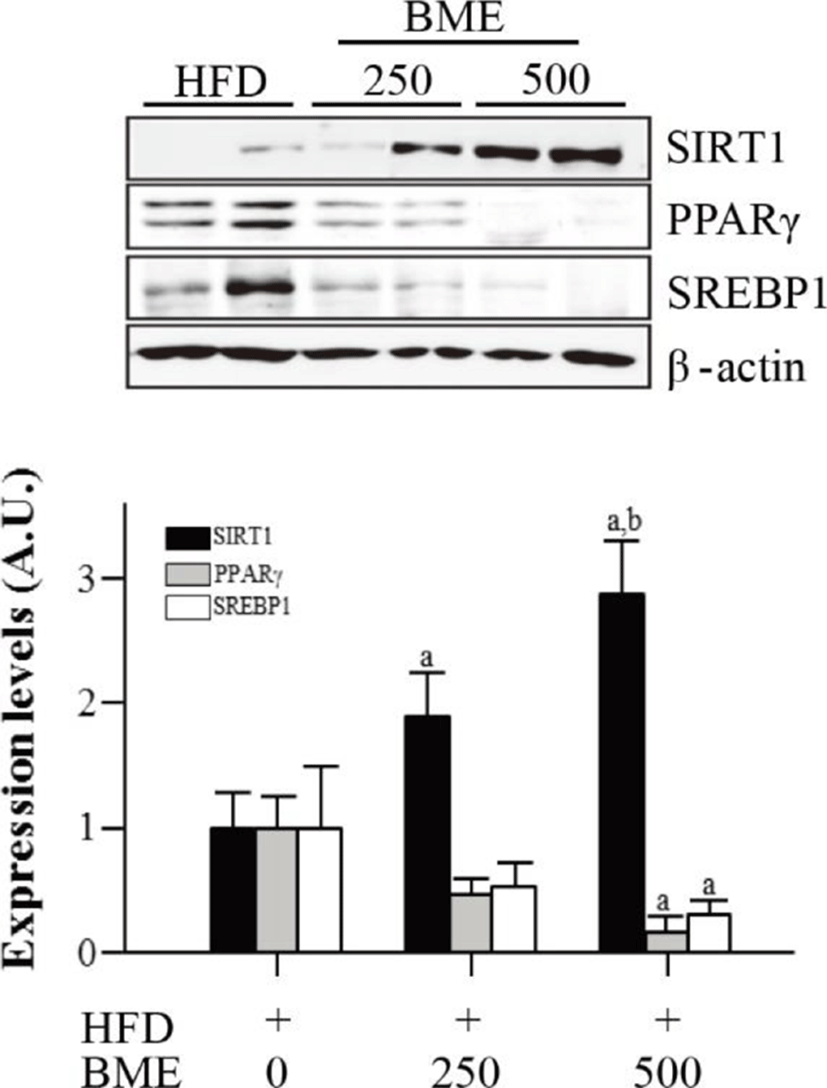
To determine whether reduced adiposity was associated with SIRT1 activation, we analyzed the expression of SIRT1 in epididymal adipose tissue isolated from the HFD group and from the BME 250 and 500 groups. The expression of SIRT1 significantly increased in the BME 250 and 500 groups compared with the HFD group. The expressions of SIRT1 target genes, PPARγ and SREBP1, decreased in the BME 250 and 500 groups compare with HFD group (Fig. 5). These data are consistent with the results obtained from 3T3-L1 adipocytes and suggest that the anti-obesity effect of BME is based on the activation of SIRT1, which inhibits adipogenesis in vivo and in vitro.
DISCUSSION
Among the several extraction methods, ethanol extraction improved the highest inhibitory activities of α-glucosidase and pancreatic lipase of BME (Moon et al., 2015). In many studies, BME has been reported to have anti-obesity effects in both rodents and humans. Bitter melon (0.75% of diet) supplementation significantly prevented body weight gain and visceral fat mass accumulation in rats fed a HFD. This weight reduction may be a result of increased fatty acid oxidation, which ultimately facilitates weight reduction (Chen & Li, 2005). In another study, bitter melon significantly decreased the weights of epididymal WAT, visceral fat, and adipose leptin and resistin mRNA levels in C57BL/6J mice fed a HFD (Shih et al., 2008). A recent study also showed that BME seed oil supplementation reduced body weight and fat mass in mice fed a HFD (Chen et al., 2012).
The improved anti-obesity effects of BME demonstrate that the polyphenol-extracted active component of bitter melon remains more stable in ethanol extract than in water extract. The BME show influence over pancreatic beta cells in streptozotocin-induced type 2 diabetic rats (Hafizur et al., 2011). Therefore, we need anticipatory and regulatory gene analyses of the anti-obesity effect of BME.
The activation of SIRT1 has been proposed as a therapeutic target for treating obesity and related metabolic disorders (Mouchiroud et al., 2013). In adipose tissue, SIRT1 regulates lipolysis and inhibits inflammation by inhibiting PPARγ and NF-κB activity (Picard et al., 2004; Pfluger et al., 2008; Mayoral et al., 2015). Another regulator, AMPK, is known to stimulate energy-generating pathways such as fatty acid oxidation and inhibit energy-consuming pathways such as lipogenesis by directly phosphorylating the key metabolic enzymes and subsequently regulating gene expression (Srivastava et al., 2012).
PPARγ is a critical transcription factor in adipocyte differentiation that stimulates the expression of the genes necessary for adipogenesis. SIRT1 has been shown to repress PPARγ by docking to the negative cofactors of the nuclear receptor and subsequently down regulating certain genes (Picard et al., 2004). Moreover, in differentiated adipose cells, the upregulation of SIRT1 leads to decreased fat storage and increased lipolysis (Baur et al., 2006; Lagouge et al., 2006). BME activated SIRT1 and reduced target genes such as PPARγ and SREBP1 in both pre-adipocytes and fully differentiated adipocytes. The activation of SIRT1 led to the suppression of 3T3-L1 differentiation, which may have prevented weight gain in the HFD-induced obese mice. The activation of SIRT1 and its target genes was also observed in the epididymal adipose tissue of the HFD mice supplemented with BME. Therefore, the anti-obesity effect of BME can be ascribed partially to the activation of SIRT1. To elucidate the anti-obesity mechanism of BME further, we examined AMPK activation because AMPK is known to be involved in the regulation of adipogenesis and the activation of AMPK suppresses adipogenesis (Han et al., 2011). AMPK may play a beneficial role in the prevention of metabolic diseases, including type 2 diabetes, obesity and cancer. It is also involved in the maintenance of lipid and cholesterol homeostasis and stimulates the oxidation of fatty acids in the mitochondria for lipid utilization (Hardie, 2008). In the present study, BME stimulated AMPK phosphorylation both in pre-adipocytes and in fully differentiated adipocytes. AMPK activation also suppresses the expressions of PPARγ, FAS, and aP2 in 3T3-L1 adipocytes and inhibits the adipogenesis of 3T3-L1 cells (Vingtdeux et al., 2011). Here, we found that BME suppressed the expression of PPARγ. Therefore, the suppression of PPARγ expression via BME treatment may be due to AMPK activation. Together, these results suggest that the activation of AMPK is also involved in the BME-mediated anti-obesity effect. In conclusion, BME may be a useful natural anti-obesity agent that reduces the risk for metabolic disorders.